


Introduction
Catatonia is a life-threatening psychomotor syndrome observed in psychiatric disorders, such as psychotic or mood disorders, alongside other medical conditions [Reference Rogers, Zandi and David1]. It is common in adult psychiatric wards, with a prevalence ranging from 9 to 30% [Reference Rogers, Zandi and David1]. In children and young adults, prevalence varies from 0.6 to 17% [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. Moreover, its prevalence among neurodevelopmental disorders is increasingly recognized [Reference Moore, Amatya, Chu and Besterman3, Reference Absoud and Malik4], and imaging studies point to anomalies in brain development (i.e. deviation of sulcation and gyrification indexes) [Reference Walther, Nadesalingam, Nuoffer, Kyrou, Wüthrich and Lefebvre5, Reference Moyal, Haroche, Attali, Dadi, Raoelison and Le Berre6] as a critical risk factor for its emergence in pediatric and adult populations [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. By proxy, genetic anomalies reported in neurodevelopmental conditions have also been identified in catatonia with neurodevelopmental features [Reference Shillington, Zappia, White, Fosdick, Erickson and Lamy7] and in pediatric populations [Reference Raffin, Consoli, Giannitelli, Philippe, Keren and Bodeau8]. In addition, these neurodevelopmentally associated-catatonia tend to exhibit heightened complexity in identification, chronicity, and resistance to treatment [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2, Reference Benarous, Raffin, Ferrafiat, Consoli and Cohen9, Reference Dhossche10]. Indeed, besides the DSM-5 criteria for catatonia (i.e. catalepsy, stupor, waxy flexibility, agitation, mutism, negativism, posturing, mannerisms, stereotypies, grimacing and echophenomena [11]), the onset of incontinence, regression in acquisitions or worsening of pre-existing symptoms such as stereotypies and self-injurious behaviors are frequent features of neurodevelopmentally associated-catatonia [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. Notably, the established diagnostic tool for catatonia, the Bush-Francis Catatonia Rating Scale (BFCRS) [Reference Bush, Fink, Petrides, Dowling and Francis12], lacks consideration of specific clinical signs often observed in these cases, such as functional regression [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. Within this neurodevelopmental context, the catatonic episodes tend to be chronic, lasting more than 12 weeks [Reference Ungvari, Leung, Wong and Lau13] and show suboptimal response to first-line treatments, lorazepam and electroconvulsive therapy (ECT) [Reference Benarous, Raffin, Ferrafiat, Consoli and Cohen9, Reference Rogers, Oldham, Fricchione, Northoff, Ellen Wilson and Mann14], due to poor drug tolerance but also because the physical condition limits access to ECT [Reference Smith, Baldwin, York, Anderson, McGonigle and Vandekar15, Reference Kolevzon, Delaby, Berry-Kravis, Buxbaum and Betancur16]. Identifying and treating catatonic episodes linked to genetic anomalies is, therefore, a significant healthcare problem that needs to be addressed.
In addition, genetic abnormalities offer valuable insights into the underlying pathophysiology [Reference Moore, Amatya, Chu and Besterman3, Reference Hirjak, Sartorius, Kubera and Wolf17]. Genome-wide association studies (GWAS) conducted on schizophrenia, bipolar disorder, and autism spectrum disorder have pinpointed various genetic polymorphisms linked to these conditions [18]. Some of these genes significantly influence neurotransmitter signaling, brain development, and synaptic function. An initial GWAS study involving 119 catatonia patients failed to unveil any specific single nucleotide polymorphism (SNPs) tied to catatonia [Reference Wilson, Sealock, Straub, Raman, Kipp and Dittus19], mainly due to the limited sample size. Much can be gained by investigating the rare variants that have been extensively reported in neurodevelopmental condition [Reference Trost, Thiruvahindrapuram, Chan, Engchuan, Higginbotham and Howe20]. A 2018 literature review [Reference Butcher, Boot, Lang, Andrade, Vorstman and McDonald-McGinn21] and a recent genetic study [Reference Shillington, Zappia, White, Fosdick, Erickson and Lamy7] highlighted a wide range of genetic abnormalities associated with catatonia with several variants implicated in the GABA/glutamate pathway. Excitation/inhibition (E/I) imbalance is a strong hypothesis in the pathophysiology of catatonia that is supported by the efficacy of GABAA agonists (such as lorazepam) and neuroimaging studies [Reference Hirjak, Kubera, Wolf and Northoff22, Reference Walther, Stegmayer, Wilson and Heckers23]. Moreover, emerging evidence suggests an autoimmune facet to catatonia [Reference Rogers, Pollak, Blackman and David24, Reference Beach, Luccarelli, Praschan, Fusunyan and Fricchione25], wherein genetic elements associated with immune system dysregulation and dysimmunity could contribute.
Hence, characterizing genetic abnormalities may provide valuable insights into catatonia’s underlying mechanisms and pave the way for more effective management strategies for these challenging, treatment-resistant catatonic episodes. In this systematic review, we aim to update the subject with a systematic review of all the genetic abnormalities reported as leading to catatonia in light of the increasing number of case reports over the last years and to perform a gene enrichment analysis to refine our understanding of the pathophysiology.
Methods
A systematic literature search was conducted for articles, including case reports, describing subjects with catatonia and genetic abnormalities. We applied the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guideline [Reference Moher, Shamseer, Clarke, Ghersi and Liberati26] and declared the review in Prospero (ID: CRD42024526691). We used PubMed and followed up on the references cited in the papers that were thus identified. The following keywords were used: (catatonia or catatonic syndrome) and (genes or genetic or variants or mutations). All relevant articles published up to July 15, 2024, were included (Figure 1). The database search was done on 15 July 2024. Papers were included in the systematic review if (a) they were published in an English-language peer-reviewed journal; (b) the study enrolled patients with catatonia; (c) the study described one or more cases of patients with catatonia and genetic anomalies. We excluded reviews and case series that did not provide data on individual patients. Articles in line with Wernicke–Kleist–Leonhard catatonia (N = 29) were also excluded from the analysis as the definition does not fit the DSM-5 catatonia criteria [11]. Following PRISMA guidelines, two different reviewers (MM and WG) searched, selected, and included articles. In case of disagreement, the other authors had to make the decision.
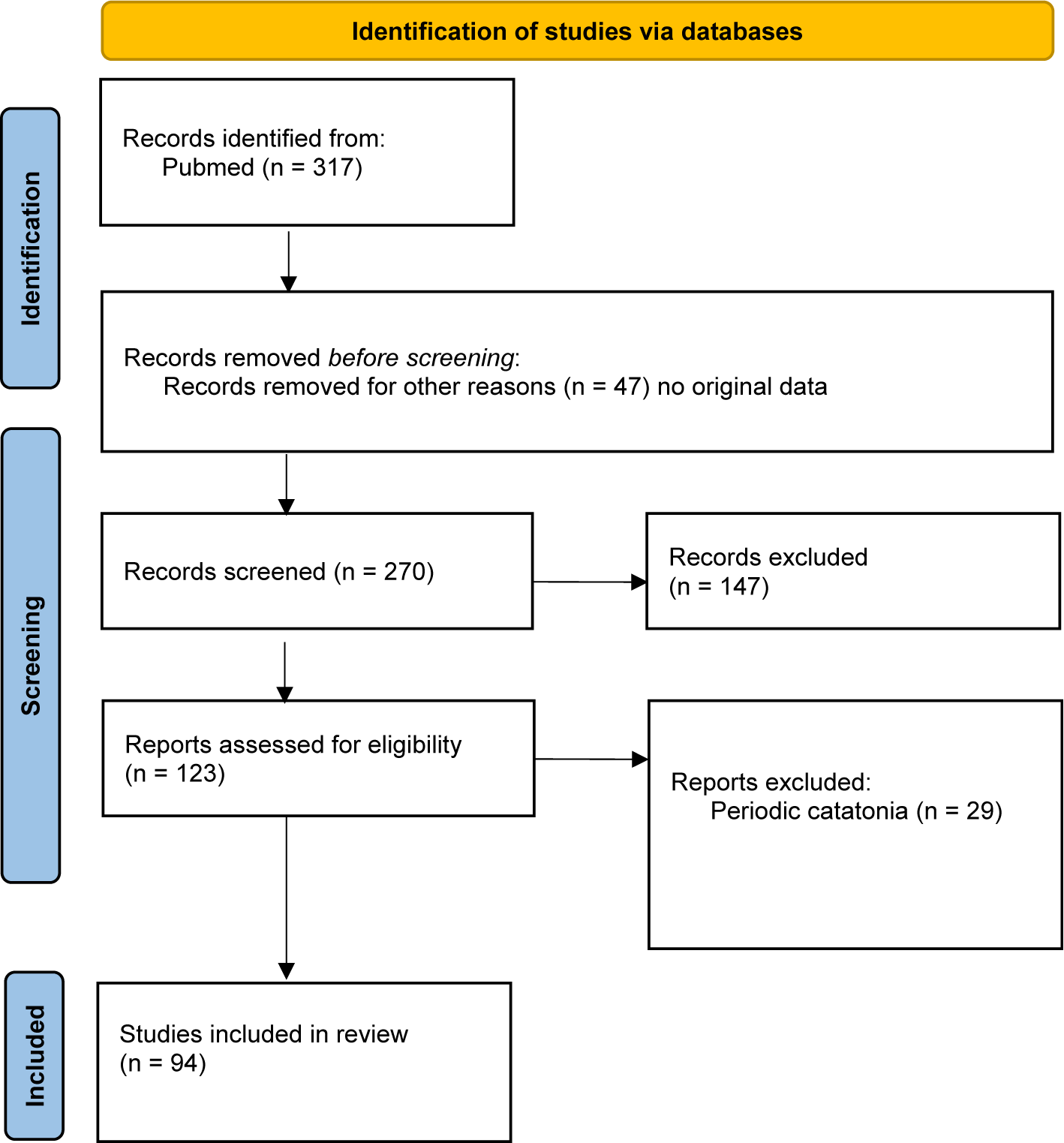
Figure 1. Flow chart.
For each report, we collected the following information: age, underlying pathology, episode characteristics, treatment used, and the catatonia diagnostic scale (Table 1). All data were extracted on 20 July 2024.
Table 1. Genetic abnormalities reported in catatonia
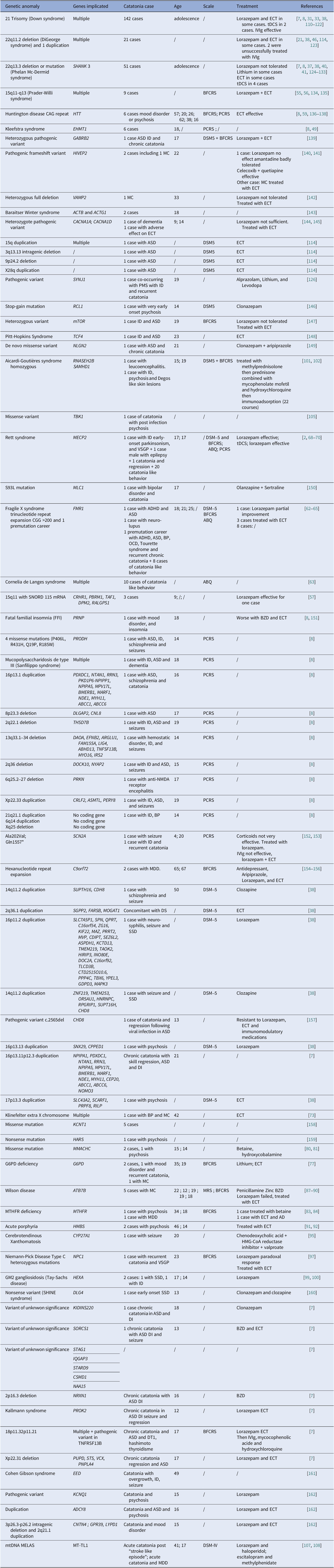
Note: With ‘/’= not available or not relevant.
Abbreviations: ABQ, attenuated behavior questionnaire; ADHD, attention deficit hyperactive disorder; ASD, autism spectrum disorder; BFCRS, Bush Francis catatonia rating scale; BP, bipolar disorder; BZD, benzodiazepines; DS, Down syndrome; ECT, electroconvulsive therapy; ID, intellectual disability; IVIg, intravenous immunoglobulins; MC, malignant catatonia; MDD, major depressive disorder; MRS, modified Rogers scale; OCD, obsessional compulsive disorder; PCRS, pediatric catatonia rating scale; SSD, schizophrenia spectrum disorder; tDCS, transcranial direct current stimulation; VSGP, vertical supranuclear gaze palsy.
We used the PhenoGram software [Reference Wolfe, Dudek, Ritchie and Pendergrass27] (http://visualization.ritchielab.org/phenograms/plot) to represent graphically the genetic abnormalities linked to catatonia (Figure 2). We used the String software (http://string-db.org) for a map of interaction (Figure 3) and the Metascape software [Reference Zhou, Zhou, Pache, Chang, Khodabakhshi and Tanaseichuk28] (https://metascape.org) to identify the biological processes and the cell type enrichment (Figure 4).

Figure 2. Location of genetic variants and small deletions and duplications reported in catatonia. The main phenotypes associated with catatonia are shown in color. The name of the gene or the size of the deletion or duplication is indicated next to it. ID = intellectual disability; ASD= autism spectrum disorder. The circle corresponds to mitochondrial DNA.
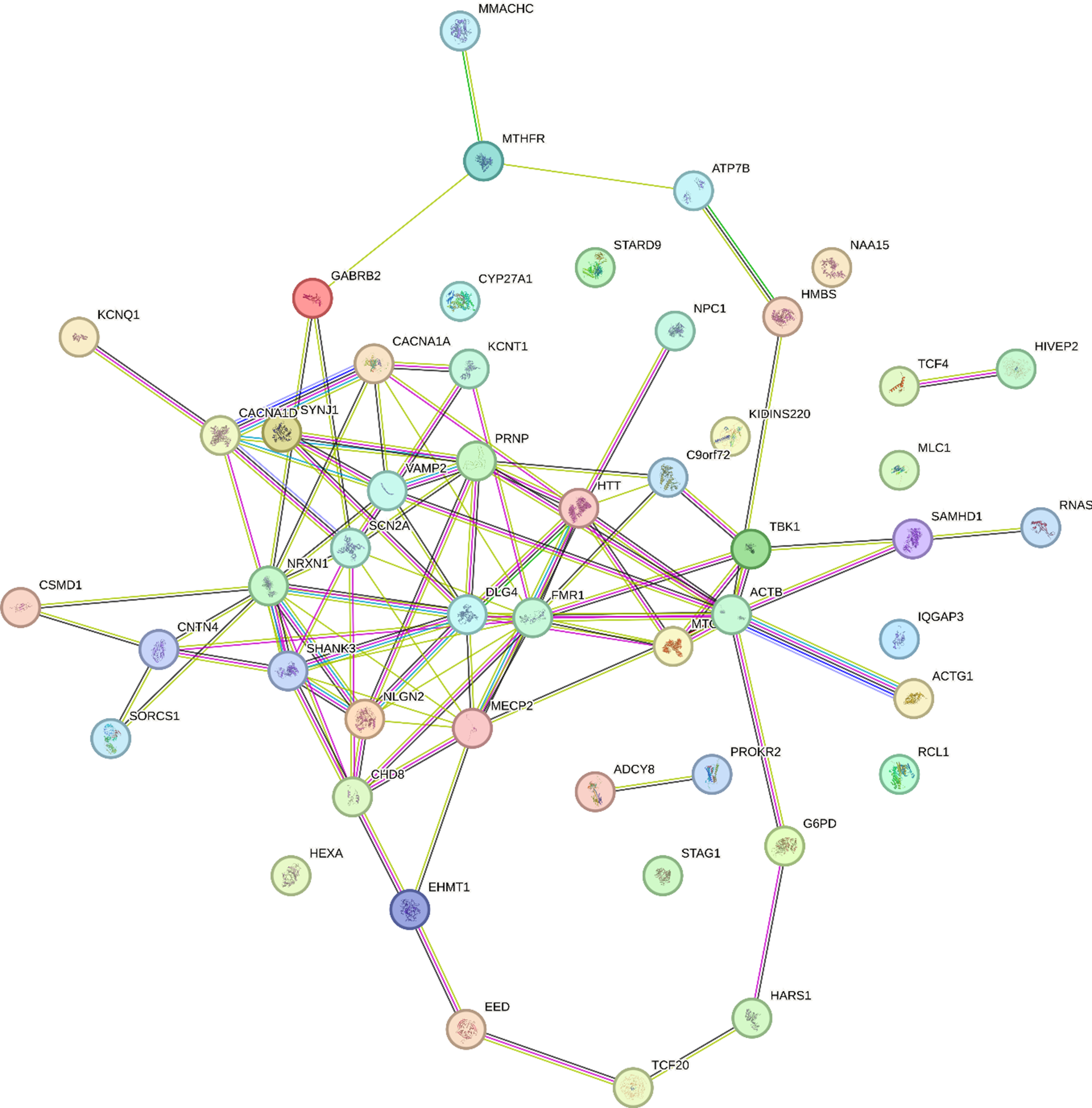
Figure 3. Gene network mapping for the 50 variants reported in catatonia.
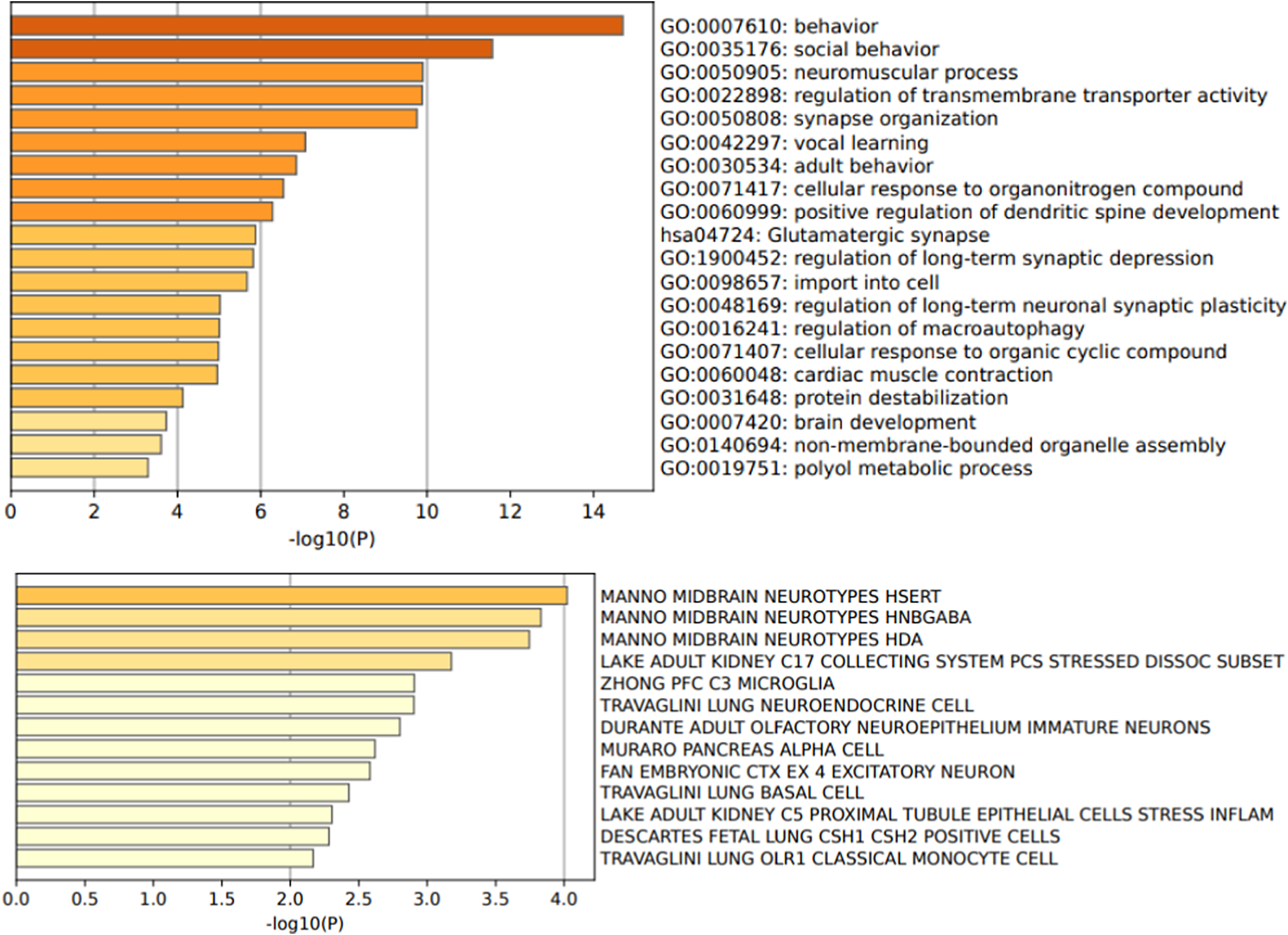
Figure 4. Gene ontology biological processes and cell type signatures of the 50 variants reported in catatonia. p-values are corrected for multiple testing and the colors represent the p-value magnitude, with darker shades indicating smaller p-values.
Results
Ninety-four articles met the criteria for our systematic review (Figure 1). This corresponds to 374 cases of catatonic syndrome and 78 different genetic abnormalities, as described in Table 1 and Figure 2. Below, we extensively describe the conditions where multiple cases of catatonia have been reported
Down syndrome: trisomy 21
Patients with trisomy 21, or Down’s syndrome, which has an incidence of 1 in 800 births [Reference Bull29], are at risk of developing a catatonic syndrome. The risk of having a catatonic episode, which is often chronic, occurs during adolescence and is generally accompanied by behavioral regression [Reference Rosso, Fremion, Santoro, Oreskovic, Chitnis and Skotko30, Reference Smith, Baldwin, Lim and Luccarelli31]. This behavioral regression, known as Down syndrome regression disorder, has been increasingly described in the literature in recent years, and a recent international experts consensus has established diagnostic criteria, including a criterion for catatonia [Reference Santoro, Patel, Kammeyer, Filipink, Gombolay and Cardinale32]. Some studies highlight the effectiveness of immunomodulatory treatments, e.g. intravenous immunoglobulin (IVIg) infusions, which are effective in 85% of cases [Reference Santoro, Spinazzi, Filipink, Hayati-Rezvan, Kammeyer and Patel33, Reference Santoro, Jafarpour, Khoshnood, Boyd, Vogel and Nguyen34]. This robust IVIg response rate is intricately linked to the well-documented autoimmune context inherent to Down syndrome [Reference Malle, Patel, Martin-Fernandez, Stewart, Philippot and Buta35]. Patients with Down syndrome regression disorder exhibit a heightened prevalence of autoimmune conditions compared to those with Down syndrome without regression, as evidenced by elevated inflammatory markers in their bloodstream [Reference Santoro, Partridge, Tanna, Pagarkar, Khoshnood and Rehmani36]. A recent study has detected 365 auto-antibodies in the plasma of individuals with Down syndrome, some targeting the central nervous and immune system [Reference Malle, Patel, Martin-Fernandez, Stewart, Philippot and Buta35]. This increased vulnerability to catatonia in these patients reinforces the hypothesis of brain inflammation as a potential causative factor in catatonia [Reference Rogers, Pollak, Blackman and David24, Reference Beach, Luccarelli, Praschan, Fusunyan and Fricchione25].
Phelan-McDermid syndrome: SHANK3 gene
Phelan-McDermid syndrome is a rare condition characterized by deletion or mutation within the SHANK3 gene in chromosome region 22q13.33. The prevalence of this syndrome is currently unknown. However, it is estimated that approximately 0.5% to 1% of ASD cases with intellectual disability are caused by PMS [Reference Kolevzon, Delaby, Berry-Kravis, Buxbaum and Betancur16]. The prevalence of catatonia within this syndrome reaches 53% [Reference Kohlenberg, Trelles, McLarney, Betancur, Thurm and Kolevzon37]. Fifty-one cases of Phelan-McDermid syndrome with catatonia have been reported in the literature see Table 1. Haploinsufficiency of SHANK3 appears to be a risk factor for catatonic syndrome [Reference Breckpot, Vercruyssen, Weyts, Vandevoort, D’Haenens and Van Buggenhout38]. SHANK3 encodes a scaffolding protein of the postsynaptic density of glutamatergic excitatory synapses. Deficiency of this protein causes hypofunction of NMDA receptors (NMDAR) [Reference Duffney, Wei, Cheng, Liu, Smith and Kittler39]. The Phelan-McDermid syndrome with catatonia model further supports the overarching hypothesis of glutamatergic system hypofunctionality in catatonia, a concept advanced by several authors, especially concerning the intricate balance between GABA and glutamate neurotransmitters [Reference Walther, Stegmayer, Wilson and Heckers23]. Furthermore, it is essential to note that all episodes of catatonia noted within Phelan-McDermid syndrome exhibit a chronic course (>12 weeks) and limited response to conventional therapeutic approaches. In this syndrome, benzodiazepines such as lorazepam can increase impulsivity, psychomotor excitation, confusion, and insomnia, limiting the use of this treatment [Reference Kolevzon, Delaby, Berry-Kravis, Buxbaum and Betancur16]. Several alternative treatments have been tried in Phelan-McDermid syndrome with catatonia, such as lithium [Reference Serret, Thümmler, Dor, Vesperini, Santos and Askenazy40] and transcranial direct current stimulation (tDCS) in 4 patients with good efficacy and safety [Reference Moyal, Plaze, Baruchet, Attali, Cravero and Raffin41]. tDCS is postulated to enhance glutamatergic synaptic function through the induction of sustained long-term potentiation, thereby fostering metaplasticity that can persist for weeks [Reference Cirillo, Di Pino, Capone, Ranieri, Florio and Todisco42].
Di George syndrome: 22q11.2 deletion
Di George syndrome, also known as 22q11.2 deletion syndrome, occurs in approximately 1 in 4,000 births, and is characterized by a deletion of variable size in the 22q11.2 region [Reference Morrow, McDonald-McGinn, Emanuel, Vermeesch and Scambler43]. A total of 21 cases of 22q11.2 deletion syndrome is reported in the literature (Table 1) is most likely underestimated, given the well-known prevalence of psychotic and mood disorders among 22q11 deletion syndrome [Reference Schneider, Debbané, Bassett, Chow, Fung and Van Den Bree44], as well as various motor abnormalities [Reference Boot, Butcher, Van Amelsvoort, Lang, Marras and Pondal45]. A less expected case of catatonia has been described in a 22q11.2 duplication [Reference Breckpot, Vercruyssen, Weyts, Vandevoort, D’Haenens and Van Buggenhout38]. The management of catatonic syndrome in these patients appears to be challenging. In Butcher and colleagues’ case series, few patients were treated with the specific treatment for catatonic syndrome, i.e. lorazepam and ECT, and the efficacy was variable. Two were unsuccessfully treated with IVIg for suspected encephalitis, and four patients had a poor outcome, including malignant catatonia [Reference Butcher, Boot, Lang, Andrade, Vorstman and McDonald-McGinn21]. The other case report shows a good response to ECT but highlights the difficulty of diagnosing catatonia with BFCRS in these patients with 22q11.2 deletion syndrome [Reference Termini, Anand, Hickox, Richter and Smith46]. The 22q11.2 region involves about 90 genes, including 46 protein-coding genes, pseudogenes, non-coding RNAs, and microRNAs [Reference Zinkstok, Boot, Bassett, Hiroi, Butcher and Vingerhoets47], in particular, the PRODH gene, which encodes a mitochondrial enzyme involved in balancing GABA/glutamate transmission [Reference Butcher, Boot, Lang, Andrade, Vorstman and McDonald-McGinn21, Reference Guna, Butcher and Bassett48]. A case of catatonia associated with a missense mutation in the PRODH gene has been reported in the literature [Reference Raffin, Consoli, Giannitelli, Philippe, Keren and Bodeau8] (Table 1). This gene may be essential in developing catatonic symptoms in patients with 22q11.2 deletion syndrome. In addition, immune dysregulation has been highlighted in this syndrome, which appears to be associated with neuropsychiatric manifestations with elevated levels of pro-inflammatory cytokines, complement activity, increased neutrophils, and blood-brain barrier dysfunction [Reference Beach, Luccarelli, Praschan, Fusunyan and Fricchione25]. This underlying neuroinflammation presents an additional avenue of risk for catatonia development, potentially synergizing with the influence of the PRODH gene.
Kleefstra syndrome: EHMT1 gene
Kleefstra syndrome emerges from the haploinsufficiency of EHMT1 due to either a deletion at 9q34.3 or pathogenic variants of EHMT1 [Reference Kleefstra, van Zelst-Stams, Nillesen, Cormier-Daire, Houge and Foulds49]. The prevalence is unknown [Reference Kleefstra, van Zelst-Stams, Nillesen, Cormier-Daire, Houge and Foulds49]. Six cases of catatonia in Kleefstra syndrome have been described in the literature (Table 1) in the context of regression of abilities. Little is known about the therapeutic approach for catatonia in these patients. EHMT1 orchestrates histone methylation and facilitates gene silencing. Alongside EHMT2, it plays a pivotal role in synaptic scaling [Reference Benevento, Iacono, Selten, Ba, Oudakker and Frega50]. A study involving excitatory cortical neurons derived from induced pluripotent stem cells obtained from Kleefstra syndrome patients exhibited EHMT1 deficiency-triggered NMDAR hyperactivity [Reference Frega, Linda, Keller, Gümüş-Akay, Mossink and Van Rhijn51]. Furthermore, insights from a Kleefstra syndrome mouse model illuminated elevated expression of specific inflammatory genes, including IL-1b, alongside an increase in activated microglial cells within the brain, thereby underscoring the presence of cerebral inflammation in these patients [Reference Yamada, Hirasawa, Nishimura, Shimura, Kogo and Fukuda52]. The predisposition to catatonia within Kleefstra syndrome might be linked to the perturbation of the E/I balance within a neuroinflammatory cerebral milieu.
Prader-Willi syndrome: 15q11-q13 region
Catatonia has been reported in Prader-Willi syndrome, associated with lack of expression of paternally inherited imprinted genes in the chromosome 15q11-q13 region generally caused by a paternal deletion or maternal disomy in with both chromosomes 15 being inherited from the mother [Reference Butler, Miller and Forster53]. Prader-Willi syndrome occurs in approximately 1 in 15,000 individuals. Neuropsychiatric features are common in this condition, with anxiety and compulsive behaviors being the most prevalent [Reference Shelkowitz, Gantz, Ridenour, Scheimann, Strong and Bohonowych54]. The use of lorazepam can be challenging in these patients with extreme obesity and obstructive sleep apnea syndrome with a higher risk of dyspnea [Reference Butler, Miller and Forster53]. ECT treatment seems to be somewhat effective [Reference Zwiebel, Rayapati and de Leon55, Reference Poser and Trutia56]. The gene region implicated in Prader-Willi syndrome also includes the brain-specific, non-coding, Small Nucleolar Ribonucleic acid C/D box 115-1(SNORD 115). SNORD 115 dysfunction, reported in 3 case reports [Reference Peter-Ross57], is hypothesized to contribute to catatonia across various neuropsychiatric disorders, including autism, schizophrenia, bipolar disorder, and major depressive disorder, as well as genetic and immune-related conditions through the regulation of five downstream genes: CRHR1, PBRM1, TAF1, DPM2, RALGPS1 and the alternative splicing of serotonin 2C receptor [Reference Peter-Ross57]. TAF1 and DPM2 provide potential clues about parkinsonism and increased creatine phosphokinase in neuroleptic malignant syndrome, while abnormalities in RALGPS1 suggest links to both anti-NMDAR antibody encephalitis and the SHANK3 gene, which is known to predispose to catatonia.
Huntington’s disease: HTT gene
Huntington’s disease, caused by an expanded CAG trinucleotide repeat in the HTT gene encoding a non-functional huntingtin protein [Reference Bates, Dorsey, Gusella, Hayden, Kay and Leavitt58], is often associated with psychiatric features in addition to motor dysfunction. It occurs approximately in one in 7,300 individuals [Reference Bates, Dorsey, Gusella, Hayden, Kay and Leavitt58]. Catatonia in Huntington’s disease is described in five cases in the literature in the context of concurrent schizophrenia and mood disorders with good response to ECT. ECT should be widely offered to patients with catatonia in Huntington’s disease. However, the diagnosis is difficult to make in patients whose psychiatric manifestations precede their motor symptoms by several years [Reference Mowafi and Millard59].
X-linked syndrome
Fragile X syndrome: FMR1 gene
Fragile X syndrome is related to a number of CGG trinucleotide repeats >200 in the FMR1 gene’s promoter region. The prevalence of the full Fragile X syndrome mutation in the general population is estimated to be approximately 1 in 5,000 males and between 1 in 4,000 and 1 in 8,000 females [Reference Hagerman, Berry-Kravis, Hazlett, Bailey, Moine and Kooy60]. Expansions between 55 and 200 repeats are defined as a premutation, and its relationship with neuropsychiatric symptoms is discussed [Reference Tassanakijpanich, Hagerman and Worachotekamjorn61, Reference Keshtkarjahromi, Palvadi, Shah, Dempsey and Tonarelli62]. A case series described eight patients with Fragile X syndrome with “catatonia-like behaviour” scored by the attenuated behaviour questionnaire (ABQ) [Reference Bell, Oliver, Wittkowski, Moss and Hare63]. Unfortunately, treatment was not described in this study. In addition, the ABQ scale used, which was developed to describe patients with ASD and catatonic symptoms, seems to overestimate the prevalence of catatonia and, to be less specific, with correlations with depression and repetitive/restrictive behavior. Moreover, a case of Fragile X syndrome and a neuro-lupus with catatonia is reported with a good response to ECT [Reference Baroud, Bond, Luccarelli, Olusunmade, Henry and Abrams64]. A male with a premutation has been described alongside ASD and ADHD associated with catatonia and a good response to ECT [Reference iIndah, Schneider, Ghaziuddin, Seritan and Hagerman65]. ECT appears to be well tolerated and effective in Fragile X syndrome or premutation careers – the FMR1 expansion results in reduced levels of Fragile X mental retardation protein (FMRP). The absence of FMRP leads to the downregulation of GABA-A and B receptors and the upregulation of glutamate receptors. This disruption in the balance of GABA/glutamate levels could likely predispose these patients to catatonia [Reference iIndah, Schneider, Ghaziuddin, Seritan and Hagerman65].
Rett syndrome: MECP2 gene
Rett syndrome results from a methyl CpG binding protein 2 (MECP2) gene mutation. This disorder predominantly impacts girls, accounting for 95 to 97% of cases and has a prevalence of fewer than 1 in 200,000 individuals [Reference Gold, Percy, Neul, Cobb, Pozzo-Miller and Issar66]. Its hallmark features encompass developmental regression coupled with movement disorders, notably hand stereotypies [Reference Temudo, Ramos, Dias, Barbot, Vieira and Moreira67]. Within Rett syndrome, an array of additional movement disorders has been documented, including a rigid akinetic syndrome prevalent in older patients. One case of catatonia in a primary school-aged girl with Rett syndrome was associated with new-onset incontinence in addition to posturing, waxy flexibility, rigidity, staring and grimacing and was resolved with 8mg of lorazepam [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. Catatonia has been described in two 17-year-old males with MECP2 deficiency [Reference Pollini, Galosi, Nardecchia, Musacchia, Castello and Nigro68, Reference Adarsh, Sharma, Sharma, Swer and Singh69]. In males carrying MECP2 variants, intellectual deficiency is associated with parkinsonism features [Reference Pollini, Galosi, Nardecchia, Musacchia, Castello and Nigro68]. One presented with a catatonic episode resolved with lorazepam, alongside a vertical supranuclear gaze palsy [Reference Pollini, Galosi, Nardecchia, Musacchia, Castello and Nigro68]; the other had recurrent lorazepam-resistant catatonia and was successfully treated with tDCS [Reference Adarsh, Sharma, Sharma, Swer and Singh69]. A noteworthy conjecture arises that several individuals with Rett syndrome and presenting parkinsonism features might be misdiagnosed and potentially could be suffering from catatonia. Using the ABQ scale, 32 patients diagnosed with Rett syndrome were evaluated [Reference Amoako and Hare70]. Of this group, 20 exhibited catatonia-like behaviors. Rett syndrome is primarily a synaptic disorder, and MECP2 deficiency impacts excitatory and inhibitory synapses, leading to an elevated E/I ratio in animal model [Reference Banerjee, Miller, Li, Sur and Kaufmann71].
Klinefelter syndrome: extra X chromosome
Klinefelter’s syndrome arises from the presence of one or more additional X chromosomes (i.e. 47XXY). Its incidence is approximately 1 in 750 births [Reference Blackburn, Ramakrishnan, Graham, Bambang, Sriranglingam and Senniappan72]. A case of catatonia is described in a 42-year-old patient with Klinefelter’s syndrome and bipolar disorder [Reference Barnardo, Pathmanaban, Dawood and Anderson73]. The presentation was rather severe, with malignant catatonia coupled with respiratory failure and effectively treated with ECT. Association between the extra X chromosome and psychiatric disorders has long been known with, in particular, an increased risk of psychotic disorders [Reference Boks, De Vette, Sommer, Van Rijn, Giltay and Swaab74]. However, very few cases of Klinefelter syndrome with catatonia have been reported, probably due to the underdiagnosis of both Klinefelter syndrome and catatonia.
Inborn errors of metabolism
Inborn errors of metabolism are linked to genetic mutations, resulting in deficiencies in metabolic enzymes and leading to the accumulation or reduced excretion of protein, carbohydrates, and lipids. These diseases are most often diagnosed in early childhood, although milder forms occurring in adolescence and adulthood may be marked by psychiatric manifestations [Reference Van De Burgt, Van Doesum, Grevink, Van Niele, De Koning and Leibold75]. Inborn errors of metabolism are individually rare, however, more than 1,000 types have been identified, with a combined prevalence of approximately 1 in 800 to 1 in 1,000 individuals [Reference Van De Burgt, Van Doesum, Grevink, Van Niele, De Koning and Leibold75]. Lahutte and colleagues have compiled a list of inborn errors of metabolism likely associated with catatonia [Reference Lahutte, Cornic, Bonnot, Consoli, An-Gourfinkel and Amoura76]. We present inborn errors of metabolism and catatonia cases documented in the literature (Table 1). Two cases of G6PD deficiency were associated with catatonia [Reference Raj, Chism, Minckler and Denysenko77] and, in particular, one with malignant catatonia requiring ECT. G6PD deficiency has been associated with psychiatric disorders, notably psychosis and mood disorders [Reference Gandar, Scott and Warren78]. It is relatively common, with an estimated global prevalence of over 500 million individuals [Reference Luzzatto, Ally and Notaro79]. Among treatable diseases, two cases of cobalamin C deficiency leading to hyperhomocysteinemia presented with catatonia in two girls of 10 and 15 years old [Reference Engel, Rashid, Beshri and Ananth80, Reference Ben-Omran, Wong, Blaser and Feigenbaum81]. In both cases, catatonia regressed with the specific treatment of hydroxocobalamin, betaine and carnitine. Cobalamin C deficiency occurs in approximately 1 in 100,000 live births [Reference Martinelli, Deodato and Dionisi-Vici82]. In addition, 2 cases of MTHFR deficiency also led to hyperhomocysteinemia in two girls, 34 and 18 years old. One was treated with betaine, yielding limited effectiveness, while the other underwent ECT [Reference Lossos, Teltsh, Milman, Meiner, Rozen and Leclerc83, Reference Ezer, Karakasli, Gurel, Ayhan and Ulusahin84]. MTHFR deficiency is the most frequent folate metabolic disorder although the incidence is very rare( <1/400,000) [Reference Froese, Huemer, Suormala, Burda, Coelho and Guéant85]. Regarding Wilson’s disease, with an estimated incidence of ~1 per 7,000 individuals [Reference Czlonkowska, Litwin, Dusek, Ferenci, Lutsenko and Medici86], five cases of catatonia have been reported, including one marked by malignant catatonia [Reference Davis and Borde87–Reference Nayak, Shetageri, Bhogale, Patil, Chate and Chattopadhyay90]. These cases raised diagnostic challenges, and in four cases, identifying the Kayser–Fleischer ring facilitated accurate diagnosis and the proposal of targeted treatments. Indeed, cases were effectively treated with the specific therapy based on penicillamine and zinc in addition to a benzodiazepine, with one case necessitating ECT. In addition, two cases of acute porphyria, with catatonia alongside psychosis exhibited significant improvements with ECT [Reference Santosh and Malhotra91, Reference Vitória-Silva and Mota92]. Acute porphyria has an estimated incidence of 10 per million [Reference Stein, Badminton and Rees93]. A case of cerebrotendinous xanthomatosis, incidence 1 in 50,000 [Reference Nie, Chen, Cao and Zhang94], and catatonia was effectively treated with the specific treatment of chenodeoxycholic acid and HMG-CoA reductase inhibitor coupled with valproate for seizure control [Reference Yadav, Singh, Mishra, Joshi, R N C and Sharda95]. A case of Niemann-Pick type C disease, incidence 0.82/100,000 [Reference Vanier96], is reported in a 23-year-old boy who exhibited recurrent catatonia alongside neurological manifestations like vertical supranuclear gaze palsy [Reference van Verseveld, Koens, de Koning, Derikx and van Waarde97]. Finally, two cases of GM2 gangliosidosis, also known as Tay–Sachs disease, incidence of one in 320,000 births [Reference Lew, Burnett, Proos and Delatycki98], were reported, and catatonia in these cases was alleviated with lorazepam [Reference Rosebush, MacQueen, Clarke, Callahan, Strasberg and Mazurek99, Reference Saleh100].
Interferonopathies
Catatonia has been described in two patients with Aicardi–Goutières syndrome [Reference Ayrolles, Ellul, Renaldo, Boespflug-Tanguy, Delorme and Drunat101, Reference Andrés, Arteche-López, Granado and Llorente102]. Aicardi-Goutières syndrome is an autosomal recessive encephalopathy within the type 1 interferonopathies characterized by increased type 1 interferon (IFN) signaling[Reference Crow and Manel103] with an incidence less than 0.7600/100,000 live births [Reference Liu and Ying104]. Both cases presented with neurodevelopmental delay associated with bilateral basal ganglia calcifications on CT scan. One had typical skin lesions. In one case, the catatonic syndrome was successfully treated with immunoadsorption (22 treatments over 8 weeks) [Reference Ayrolles, Ellul, Renaldo, Boespflug-Tanguy, Delorme and Drunat101]. Another case of catatonia was described in a woman with post-infectious psychosis and a TBK1 variant [Reference Izu, Mooneyham, Ngo, Pleasure, Wilson and Nath105]. TBK1 upregulates type 1 IFN transcription genes. Type 1 IFN is essential as an antiviral cytokine and regulates innate and adaptive immune responses. Dysregulation of IFN signaling could play a key role in triggering the cerebral inflammation that leads to catatonia [Reference Beach, Luccarelli, Praschan, Fusunyan and Fricchione25].
Mitochondrial DNA mutation
Catatonia has been documented in two cases involving mitochondrial DNA mutation, the MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes) syndrome, prevalence of 16 to 18/100,000 [Reference El-Hattab, Adesina, Jones and Scaglia106]. A 41-year-old woman presented with catatonia following stroke-like episodes (SLE) [Reference Montano, Simoncini, LoGerfo, Siciliano and Mancuso107], a hallmark symptom of MELAS driven by ictal activity leading to a range of neurological and psychiatric manifestations. The catatonic episode was successfully resolved with benzodiazepines. In another case, a patient with MELAS exhibited acute catatonia in the context of a major depressive disorder, which responded well to antidepressant treatment [Reference Ryu, Lee, Sung, Ko and Yoo108]. Psychiatric symptoms are prevalent in mitochondrial disorders ranging from 6 to 28%, including anxiety, mood, and psychosis disorders [Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush109], and are likely to be related to a dysfunction of the brain’s respiratory chain. Antipsychotics must be cautiously considered, both typical and atypical, as they have been shown to inhibit complex I of the respiratory chain, potentially exacerbating the symptoms [Reference Anglin, Garside, Tarnopolsky, Mazurek and Rosebush109].
Clinical and therapeutic overview
Of 353 cases with a complete clinical description, 241 cases (68%) had a chronic or recurrent presentation, and 14 cases (4%) presented with malignant catatonia. Of 311 cases with documented treatment data, 261 cases (84%) showed a poor response to lorazepam, and 135 cases (43%) were treated with ECT, in contrast to the typical 70% good response rate to lorazepam in catatonia [Reference Rogers, Zandi and David1]. Only three were reported to be treated with clozapine.
Enrichment analyses
This systematic literature review identified 50 genetic variants associated with catatonia alongside 23 small-scale duplications and deletions. In our analysis, we have excluded genes encompassed by deletions and duplications, as it is challenging to determine which subset of genes plays a significant role in catatonia. The 50 genetic variants exhibit high interconnectivity, suggesting a potential convergence of shared biological processes, as illustrated in Figure 3. The protein-protein interaction revealed pathway enrichment in postsynaptic membrane organization (p = 6.31×10−13), positive regulation of excitatory postsynaptic potential (p = 10–12), and receptor clustering (p = 5×10−12). The variants are enriched in genes involved in behavior (p = 2 × 10−15), social behavior (p = 2 × 10−12), regulation of transmembrane transporter activity (p = 3.63 × 10−11), synapse organization (p = 1.2 × 10−10) and neuromuscular process (p = 1.1 × 10−10) (Figure 4). These genes are mainly expressed in GABAergic (p = 1.5 × 10−4), serotoninergic (p = 1 × 10−4), dopaminergic (p = 2 × 10−4), and excitatory (p = 1.6 × 10−3) neurons as well as in microglial cells (p = 5 × 10−3) (Figure 4).
Discussion
In this review, we reported all the genetic anomalies associated with a catatonic syndrome described in the literature to date. This analysis included 94 articles with 374 cases of catatonia and 78 distinct genetic anomalies.
Firstly, regarding clinical presentation, this review highlights that catatonic episodes associated with genetic abnormalities exhibit a chronic course in 241 cases (68%). Additionally, typical clinical signs of catatonia may be absent, with behavioral regression often being a predominant feature. A recent review article proposes assessing a personalized score at baseline based on the BFCRS and the pediatric catatonia rating scale (PCRS) [Reference Benarous, Consoli, Raffin, Bodeau, Giannitelli and Cohen163] to facilitate diagnosis and assessment of response to treatment [Reference Hauptman, Cohen, Dhossche, Raffin, Wachtel and Ferrafiat2]. In addition, some cases of catatonia with genetic abnormalities have been described in neurodegenerative disorders such as C9orf72, associated with frontotemporal dementia, and in Huntington’s disease, reinforcing the importance of looking for genetic abnormalities also in neurodegenerative disorders in line with the known associated vulnerability between neurodevelopmental and neurodegenerative disorders [Reference Douaud, Groves, Tamnes, Westlye, Duff and Engvig164].
Concerning the therapeutic aspect, in the majority of cases (261 cases; 84%), lorazepam proved inadequate in alleviating catatonia, exhibiting reduced efficacy or poor tolerance. Electroconvulsive therapy was frequently used (135 cases; 43%) and was mostly effective but limited by its restrictive access [Reference Smith, Baldwin, York, Anderson, McGonigle and Vandekar15, Reference Charpeaud, Tremey, Courtet, Aouizerate and Llorca165]. Thus, although ECT should be widely offered [Reference Smith, Baldwin, Termini, McGonigle, Vandekar and Luccarelli166], alternative treatments must be discussed. Although only three cases of catatonia were treated with clozapine with significant improvement, clozapine appears to be well tolerated and effective in patients with neurodevelopmental disorders [Reference Thom, Wu, Ravichandran and McDougle167] and should therefore be part of the personalized care strategies considered. The efficacy of clozapine may be closely linked to the imbalance in the E/I ratio and, in particular, to the involvement of gabaergic interneurons [Reference Plevin, Mohan and Bastiampillai168, Reference Nair, McKinnon, Miners and Bastiampillai169]. Indeed, clozapine may facilitate the binding of GABA to the GABAB receptor [Reference Wu, Blichowski, Daskalakis, Wu, Liu and Cortez170]. tDCS, an easy-to-apply noninvasive brain stimulation technique, was effective and safe in 16 cases of catatonic patients, including four patients with Phelan-McDermid syndrome [Reference Moyal, Plaze, Baruchet, Attali, Cravero and Raffin41, Reference Haroche171] and two patients with Down Syndrome Regression Disorder [Reference Brunelin, Adam, Favre, Prange, Zante and Demily172]. In addition, in catatonia cases related to neuroinflammation, anti-inflammatory interventions appear promising [Reference Santoro, Jafarpour, Khoshnood, Boyd, Vogel and Nguyen34, Reference Ferrafiat, Raffin, Freri, Granata, Nardocci and Zibordi173]. Anti-inflammatory treatment needs to be offered more widely, even in the absence of autoantibodies identified by lumbar puncture or blood test, in syndromes with known inflammation such as Down Syndrome Regression Disorder [Reference Santoro, Spinazzi, Filipink, Hayati-Rezvan, Kammeyer and Patel33]. Along the same lines, specific treatment in case of treatable inborn errors of metabolism, such as Cobalamin C disorder or Wilson disease, can treat catatonia. Knowledge of genetic abnormalities can help organize timely personalized care [Reference Moyal, Plaze, Baruchet, Attali, Cravero and Raffin41, Reference Wigby, Nicolas, Carpinello, Ricciardi and Willis140]. Likewise, it is legitimate to systematically assess catatonic symptoms in genetic diseases, notably in Down syndrome and Phelan–McDermid syndrome, where its prevalence is extremely high [Reference Santoro, Patel, Kammeyer, Filipink, Gombolay and Cardinale32, Reference Kohlenberg, Trelles, McLarney, Betancur, Thurm and Kolevzon37].
In light of this literature review, genetic testing for catatonia should be considered more broadly in patients with the following criteria: the presence of a neurodevelopmental background [Reference Shillington, Zappia, White, Fosdick, Erickson and Lamy7], a chronic episode lasting more than 12 weeks, recurrent episodes, early onset of catatonia, resistance or paradoxical reaction to first-line treatment of catatonia, association with neurological signs including seizures, and the existence of malignant catatonia. Whole-exome or whole-genome sequencing should be considered the first-tier diagnostic framework, considering the high proportion of SNVs.
Although the risk of developing catatonia is high in certain genetic conditions, the onset of a catatonic episode may depend on environmental factors, such as exposure to antipsychotics [Reference Hirjak, Sartorius, Kubera and Wolf17] or the sudden discontinuation of clozapine treatment [Reference Lander, Bastiampillai and Sareen174]. Well-conducted studies are needed to identify environmental risk factors in these genetically predisposed patients to develop effective prevention strategies.
Finally, a deeper understanding of the genetic factors involved in catatonia can shed light on its pathophysiology. The protein–protein interaction, enrichment in biological processes, and the cell type signatures in the 50 genetic variants associated with catatonia reinforce the hypothesis of a dysfunction at the synaptic level, particularly in GABAergic neurons. It supports the likely central role of GABAergic interneurons in catatonia [Reference Lander, Bastiampillai and Sareen174, Reference Lefebvre, Gehrig, Nadesalingam, Nuoffer, Kyrou and Wüthrich175] in the hypothesis of E/I imbalance [Reference Walther, Stegmayer, Wilson and Heckers23]. Glutamate modulates the activity of the GABAergic interneurons through their NMDA receptors that regulate the synchronization of superficial pyramidal cells, thus ensuring a variety of cognitive processes [Reference Uhlhaas and Singer176]. A disruption of GABAergic interneurons has been highlighted in several psychiatric disorders, including ASD and schizophrenia [Reference Uhlhaas and Singer176, Reference Port, Oberman and Roberts177], both of which are associated with catatonia. This disruption may underlie the dysconnectivity between the medial prefrontal, orbitofrontal, and motor areas observed in fMRI studies of catatonia [Reference Haroche, Rogers, Plaze, Gaillard, Williams and Thomas178] and could represent a shared pathophysiological mechanism linking these disorders to catatonia. Furthermore, as interneuron-dependent prefrontal cortical maturation occurs during adolescence [Reference Caballero and Tseng179], their disruption may also explain the specific timing of catatonia onset in syndromes such as DSRD. Adolescence thus emerges as a vulnerable timeframe for catatonia. Quantitative EEG analyses could be used to provide further support for the hypothesis of disruption of GABAergic interneurons in catatonia [Reference Ahmad, Ellis, Leech, Voytek, Garces and Jones180]. Such anomalies in GABAergic function could be due to various pathological processes, contributing to the diverse clinical presentations observed in catatonia. One potential mechanism is neuro-inflammation, a phenomenon observed in several genetic diseases, notably Down’s syndrome and 22q11DS, and also reported in catatonia [Reference Rogers, Pollak, Blackman and David24, Reference Beach, Luccarelli, Praschan, Fusunyan and Fricchione25]. Autoantibodies targeting GABAergic interneurons could lead to catatonia, as it has also been observed for autoantibodies targeting NMDA receptors [Reference Espinola-Nadurille, Flores-Rivera, Rivas-Alonso, Vargas-Cañas, Fricchione and Bayliss181]. The microglial cell signature of genetic variants associated with catatonia further underscores the involvement of neuroinflammation. Based on our cell-enrichment analysis, serotonergic and dopaminergic neurons also appear to be involved. Serotonin receptors have previously been associated with catatonia, notably via SNORD 115 mRNA, which regulates alternative splicing of the serotonin 2C receptor [Reference Peter-Ross57]. Furthermore, catatonia is frequently associated with both serotonin syndrome and neuroleptic malignant syndrome, each respectively associated with an excess of serotonin and dopamine blockage [Reference Hauptman and Benjamin182].
Several methodological issues call for caution when interpreting this work. First, catatonia is not always described using the same scale, and sometimes no scale has been used. It is rather difficult to ascertain the diagnosis and severity of the syndrome. Second, we probably missed genetic abnormalities as the vast majority of genetic cases with catatonia have not been published, and there is still limited access to genetic testing in psychiatric disorders. Another form of bias stems from the limited conclusiveness of cases due to our current understanding of genetics. This understanding is influenced by variants found in other psychiatric conditions, resulting in the expectation that biological processes and cell signatures will be concentrated in usual brain pathways. In addition, given the absence of a control group, we cannot ascertain whether these genetic variations are truly enriched in catatonia specifically or if their presence merely reflects broader neurodevelopmental processes associated with catatonia. Nevertheless, these findings provide a valuable foundation for generating research hypotheses. To overcome this limitation, future studies should incorporate larger, well-controlled, and unbiased genetic analyses involving patients with catatonia.
Conclusion
This review highlighted the neurodevelopmental burden of catatonia and the clinical relevance of genetic explorations in this syndrome while also discussing the underlying pathophysiology and its implications for treatment. Our systematic description of rare disorders associated with catatonia, providing clear links between genotype and phenotype, strengthens the importance of GABAergic interneuron dysfunction. This work further points out the importance of large-scale whole-exome or whole-genome sequencing studies to identify new genetic variants and pathways associated with catatonia. Improving our knowledge of catatonia’s genetic background may allow for the development of more targeted diagnostic approaches and personalized treatment strategies.
Data availability statement
All the data are available.
Acknowledgements
We would like to thank Alexandre Gestin for his assistance with the PhenoGram software.
Financial support
Mylène Moyal received funding from the John Bost Foundation.
This work is supported by a Young Researcher grant (ANR JCJC ANR-22-CE16-0029), by a grant from Inserm and the French Ministry of Health (Inserm-AAP Messidore2022-N°9), and by a French government grant managed by the Agence Nationale de la Recherche under the France 2030 program (reference ANR-22-EXPR0013).
Competing interests
The authors have declared that there are no conflicts of interest concerning the subject of this study.






 Open access
Open access
Comments
No Comments have been published for this article.