


Background
I am honoured to be the third recipient of the Nutrition Society’s 2024 Senior Award: ‘The Blaxter Award’. Professor Sir Kenneth Lyon Blaxter FRS, (1919–1991) was a remarkable man and a widely recognised very distinguished Scientist and Past President of the Society. For those with an interest in the history of our discipline, his fascinating life and work have been described in detail by Waterlow and Armstrong 1993(Reference Lyon Blaxter1) and by John Webster in the Society Gazette (Summer 2019(Reference John2)). My interaction with him was brief at the very start of my career in the 1970s, as detailed below. At that time my interest in protein turnover and the nutritional role of protein resulted from a chance application to Professor John Waterlow for a PhD studentship with him after my first degree in Biochemistry at Cardiff in 1966. I was taken on at the MRC Tropical Metabolism Research Unit at the University of the West Indies in Jamaica and my life changed forever. There my interest in the regulation of growth was kindled by the Unit’s work on the rehabilitation of severely malnourished infants which Anne Ashworth, David Picou, John Garrow and George Alleyne and other TMRU staff under Waterlow had pioneered. Such infants with kwashiorkor, oedematous malnutrition, have little desire to eat on admission to hospital, and cannot tolerate high-protein foods, which can be lethal especially when tube-fed. It is likely that an infection coupled with micronutrient deficiency invariantly observed in such infants, was inhibiting protein synthesis in muscle as it does in infected rats(Reference Jepson, Pell and Bates3). Thus, even though the infants are wasted, dietary amino acids cannot be disposed of as tissue protein deposition and the capacity for amino acid oxidation may be insufficient to avoid very high postprandial amino acid levels in the blood in addition to the other components of refeeding syndrome. With appropriate treatment with antibiotics, electrolytes, key micronutrients and cautious feeding of maintenance energy and protein, their oedema is lost, their condition stabilises, and a ravenous appetite returns enabling the consumption of large amounts of an energy-rich diet, as long as the protein content is modest (e.g. ≈10 % of energy), and amino acid supply does not exceed the capacity for protein deposition. Their hunger and food intake falls when they achieve their expected weight for height, after which they eat and grow normally(Reference Ashworth and Millward4)
I left Jamaica in 1969 and after a 1 year post doc in the Biochemistry department at UCL, I moved to the Human Nutrition department at the London School of Hygiene, (LSHTM) in 1970 with John Waterlow after his appointment to the departmental Chair on the death of Ben Platt in 1969. Blaxter had been interested in protein metabolism(Reference Blaxter and Munro5) while at the Hannah Dairy Research Institute (1948–1965), and in 1972 was director of the Rowett Research Institute in Aberdeen where the society organised a joint symposium with the Biochemical Society on ‘The Influence of Amino Acid Supply on Polynucleotide and Protein Metabolism’ at which Peter Garlick and I gave the first two papers(Reference Garlick and Millward6,Reference Millward and Garlick7) . We both met Blaxter at the meeting. Shortly after, Blaxter decided to expand protein work at the Rowett and asked Peter Garlick and myself to join a new unit he was setting up in Aberdeen. For a variety of reasons we declined since we were well provided for, especially after Waterlow had established, through the Wellcome Trust, the Clinical Nutrition and Metabolism Unit (CNMU) at the Hospital for Tropical Diseases at St Pancras in London where all departmental experimental animal and human work subsequently occurred. This was formally opened in 1973 by Princess Alice, Countess of Athlone, (aged 90), who, in 1955 as Chancellor of the University of West Indies had opened the TMRU in Jamaica. This new unit provided some continuity for Waterlow’s work and soon became an internationally recognised centre for excellence. I often wonder how a move to Aberdeen would have turned out.
Waterlow retired in 1982, although he continued to work and publish up to just before his death in 2010. In 1992 I moved to Surrey as Professor and Departmental Chair of Human Nutrition where my research diversified up to my full retirement in 2009 as Emeritus Professor of Human Nutrition. I continued to teach, as a visiting Professor of Medical Nutrition, at St Georges University Medical School, Grenada till 2017 and at the LSHTM, UCL and at Surrey up to the present time.
Proteostasis and turnover: development of the knowledge base
What we called protein turnover is now included within the descriptor, proteostasis, an umbrella term describing all aspects of the maintenance of an intact proteome within cells, acting through a proteostasis network, (PN) (see Millward 2021(Reference Millward8) and 2024(Reference Millward9) and below for more details). The concept of proteostasis as a dynamic process was discovered by the pioneering stable isotope work of Schoenheimer, Rittenberg, and Ratner with deuterated and 15N labelled amino acids in the 1930s and summarised in Schoenheimers book in 1942(Reference Ratner10,Reference Young and Ajami11) . However, there was little progress for several decades until the exploration of protein turnover was restarted mainly with the simplifying work of John Waterlow and Joan Stephen in the 1960s. They introduced the constant intravenous infusion of [U-14C] lysine method to study the response to protein deficiency, which was, incorrectly as it turned out, believed at the time to be the cause of Kwashiorkor, i.e. oedematous childhood malnutrition. This new method allowed accurate in vivo measurements of protein turnover in the whole body and tissues(Reference Waterlow and Stephen12–Reference Waterlow15), but the methodology was laborious and progress was slow.
My own early studies of the decay rates of tracer-labelled proteins(Reference Millward16,Reference Millward17) , while highly cited and considered at the time to be biochemically elegant, proved much less practical than the precursor-product approach, so that the simplifying of this approach at the CNMU enabled a step change in the range and volume of studies in animal models. Thus tail-vein tracer infusions of U-14C tyrosine, a single large (flooding) dose of [3H] phenylalanine given iv and then by intraperitoneal injections, coupled to simple fluorimetric assays for the specific activity of free and protein-bound U-14C tyrosine and [3H] phenylalanine(Reference Jepson, Pell and Bates3,Reference Garlick and Marshall18,Reference Garlick, McNurlan and Preedy19) , allowed rates of muscle protein synthesis (MPS) to be measured in vivo accurately and relatively simply. Such rates could then be compared with the rate of changes in the muscle protein content in response to the interventions, (i.e. positive or negative growth rates of the tissue protein studied), enabling calculation of the rate of protein degradation (MPD). Thus these methods, together with a variety of other tracer methods enabled, by the end of the 1980s, a complete description of protein turnover in murine muscle during development(Reference Millward, Garlick and Stewart20,Reference Bates and Millward21) , in response to dietary and hormonal manipulations(Reference Giugliano and Millward22–Reference Brown, Van Bueren and Millward33), corticosteroids(Reference Odedra and Millward28,Reference Odedra, Dalal and Millward34,Reference Odedra, Bates and Millward35) , anabolic steroids(Reference Bates, Chew and Millward36), and endotoxemia(Reference Jepson, Pell and Bates3). Stretch-induced skeletal muscle hypertrophy was studied in an adult avian model by constant intravenous infusion of [14C]proline(Reference Laurent, Sparrow and Bates37–Reference Laurent and Millward39) and we completed our animal studies with the documentation of the extent of muscle-bone interactions in response to dietary protein and energy deficiency(Reference Yahya, Bates and Tirapegui40–Reference Yahya, Tirapegui and Bates43), which, together with our previous work allowed the first formulation of the protein stat model for growth regulation in 1995(Reference Millward44), which has recently been updated(Reference Millward9,Reference Millward45) . Finally, the application of these tracer kinetic approaches with stable isotopes (15Nglycine, [2H5]phenylalanine, [2H2,]tyrosine and 13C-1 leucine), to human studies of proteostasis and postprandial protein utilisation was developed in collaboration with Mike Rennie, Dave Halliday and Paul Pacy allowing many of the concepts developed in the murine models to be examined in human muscle(Reference Rennie, Edwards and Halliday46,Reference Millward and Smith47) , as well as in the whole body(Reference Price, Halliday and Pacy48–Reference Millward, Fereday and Gibson53), so that adaptation to varying protein intakes could be studied. This was especially important for the development of the adaptive metabolic demand model for protein requirements during maintenance(Reference Millward54).
Putting it all together
There were three elements within our Protein-Stat model of the regulation of the LBM by dietary protein(Reference Millward44,Reference Millward45) , namely the control of growth, of maintenance and of protein intake by appetite and these will be considered here separately in the description of the development of the model.
Growth
From first principles, growth control was assumed to be the regulation, by genetic determinants and functional demand, of nutrient substrate flow into energy storage, growth (defined as irreversible structural change) and metabolic consumption. Because nutrition exerts active regulation in terms of specific dietary influences on anabolic processes, especially in the case of dietary protein, it was assumed to exert both permissive and active regulation.
Hierarchical model of regulation of growth & body composition
Growth in terms of body composition and the fat-free mass can be considered to occur through bone length growth driving appended muscle growth which in turn drives increasing food intake, the main driver of growth of the visceral organs, in response to functional demand generated by nutrient intake and metabolism.
Bone lengthening occurs at rates determined by genetic programming and mediated by growth hormone, secretion of which is to a large extent sleep-dependent, by thyroid hormones and by the sex steroids during puberty, acting together with an appropriate anabolic drive deriving from dietary protein to drive endochondral ossification in the growth plate(Reference Millward55). The outcome of bone length growth is adult height, a highly polygenic trait, of which the heritability is 90 % in men and 70–80 % in women with over 12 000 SNPs currently known which account for 40 % of the phenotype(Reference Yengo, Vedantam and Marouli56).
The phenotypic appendicular adult lean mass is also an independent highly polygenic trait with a heritability of 50 % with > 1000 variants currently known(Reference Pei, Liu and Yang57). The link between bone length and muscle mass growth is mechanotransduction mechanisms, (see(Reference Millward9) for recent review). This involves both passive stretching of muscle by bone lengthening which induces muscle myofibre lengthening by the addition of new sarcomeres, and muscle contraction and internal force development associated with muscle work against the forces of gravity which increases myofibre and whole muscle cross sectional area (CSA) and muscle weight. These forces acting on muscle ensure growth occurs so that muscle strength, which varies directly with CSA, is appropriate for the demands made on it by gravity, which varies with body weight (∝ length3). This means that muscle weight, a function of length × CSA, should vary with the 4th power of bone length. This is what we observed for the appendicular muscle-bone pair of the gastrocnemius and tibia in the growing rat(Reference Yahya and Millward42), a relationship also observed in human muscle growth with quadriceps strength varying as height3 (i.e. weight varying as height4) in boys and girls(Reference Parker, Round and Sacco58)
Muscle growth as ‘bag’ enlargement and filling
Muscle growth involves myofibre hypertrophy through both myofibrillar protein deposition within the expanding myofibres as well as remodelling of their encasing extracellular matrix, (ECM) of connective tissue. The ECM is most important as a structure which allows force generated within the myofibre to be transmitted to its bone levers either directly onto bone or indirectly via aponeuroses and tendons. It comprises a complex connective tissue structure which encases each myofibre, (the endomysium or basal lamina), the fascicles (bundles of myofibres encased in the perimysium), and the entire muscle (epimysium). Thus the ECM defines myofibre and muscle volume and carries the majority of muscle passive load(Reference Jorgenson, Phillips and Hornberger59) and during growth its remodelling must be complex.
The Protein-Stat deals with this with a simple structural model for muscle growth and maintenance, originally introduced in 1988(Reference Millward, Barth and Schlimme25), which we called the ‘bag’ theory of muscle growth. This model was based on the simple concept that muscle could be conceived as a system of concentric connective tissue bags, its volume and consequent mass limited by the connective tissue extracellular matrix in terms of the epi, peri and endomysial sheaths, and was a speculative idea. It allowed muscle growth to be conveniently described in terms of ‘bag’ enlargement, through ECM remodelling, and ‘bag’ filling in terms of protein deposition within the myofibre. It provided a potential mechanism for the stability of muscle mass throughout the life cycle and prior to the onset of sarcopenia. Specifically during the diurnal cycle of post absorptive losses and postprandial gains of body protein(Reference Price, Halliday and Pacy48,Reference Millward, Price and Pacy60) , losses and gains of muscle could be identified as ‘bag’ emptying and refilling until a ‘bag-full’ signal inhibited postprandial protein synthesis, terminating protein deposition in the myofibre when the phenotypic, regulated size of skeletal muscle mass was achieved. The evidence for this and some potential mechanisms for the ‘bag full’ signal is discussed below.
The myonuclear domain and the role of muscle stem cells in muscle growth
The unique feature of muscle structure is that the contracting element, the myofibers are a syncytium, a multinucleate cell containing hundreds or thousands of post-mitotic myonuclei beneath the sarcolemma, with no discernible structural compartmentation between them(Reference Bagley, Denes and Mccarthy61). Thus the myonuclear domain (MND) is a necessary concept as a volume of cytoplasm within which proteostasis is controlled by a single nucleus with units of the protein/DNA ratio. The concept remains valid even with the recent suggestion that in some but not all mouse muscles there may be endoreplication of a very small fraction of myonuclei resulting in some polyploidy(Reference Borowik, Davidyan and Peelor62). Polyploidy is a strategy which allows a nucleus to manage a larger volume of cytoplasm, and is best understood for the liver(Reference Matsumoto, Wakefield and Tarlow63). The roles of myonuclei in adult skeletal muscle health and function has recently been reviewed(Reference Borowik, Murach and Miller64).
Our measurements in the adult fowl showed a fibre-type specific MND-protein turnover relationship with muscles with mainly slow oxidative fibres exhibiting faster turnover rates and smaller protein/DNA ratios compared with fast glycolytic muscles (Figure 1). However, each myonucleus in these two muscle types managed the same amount of protein synthesis(Reference Laurent, Sparrow and Bates37), a relationship maintained after the stretch-induced hypertrophy of the muscle(Reference Laurent, Sparrow and Bates65). In the adult rat similar fibre-type specific MND size and turnover relationships are observed(Reference Waterlow, Garlick and Millward66,Reference Millward, Bates and Laurent67) . However during growth and development of the mainly fast type II muscles, (gastrocnemius and quadriceps), there was a complicated two-phase relationship between turnover and growth in MND size(Reference Millward, Garlick and Stewart20). The very high turnover at weaning, and in early post natal life, most likely related to intense muscle fibre remodelling(Reference Tamaki, Akatsuka and Yoshimura68), fell in concert with some growth in apparent MND-size. For growth during adult life, which occurs in the male rat because of a lack of fusion of the growth plate in bone, turnover was low and stable and MND size growth occurred mediated by a > 50 % increase in transcriptional activity and translation, (protein synthesis per unit DNA), associated with an increase in myonuclear ribosomal capacity: i.e. the RNA/DNA ratio. This response enabled the MND size to increase without any further reduction in turnover. Clearly, this is a different strategy than observed during the stetch-induced hypertrophy in the adult fowl(Reference Laurent and Millward39,Reference Laurent, Sparrow and Bates65) .
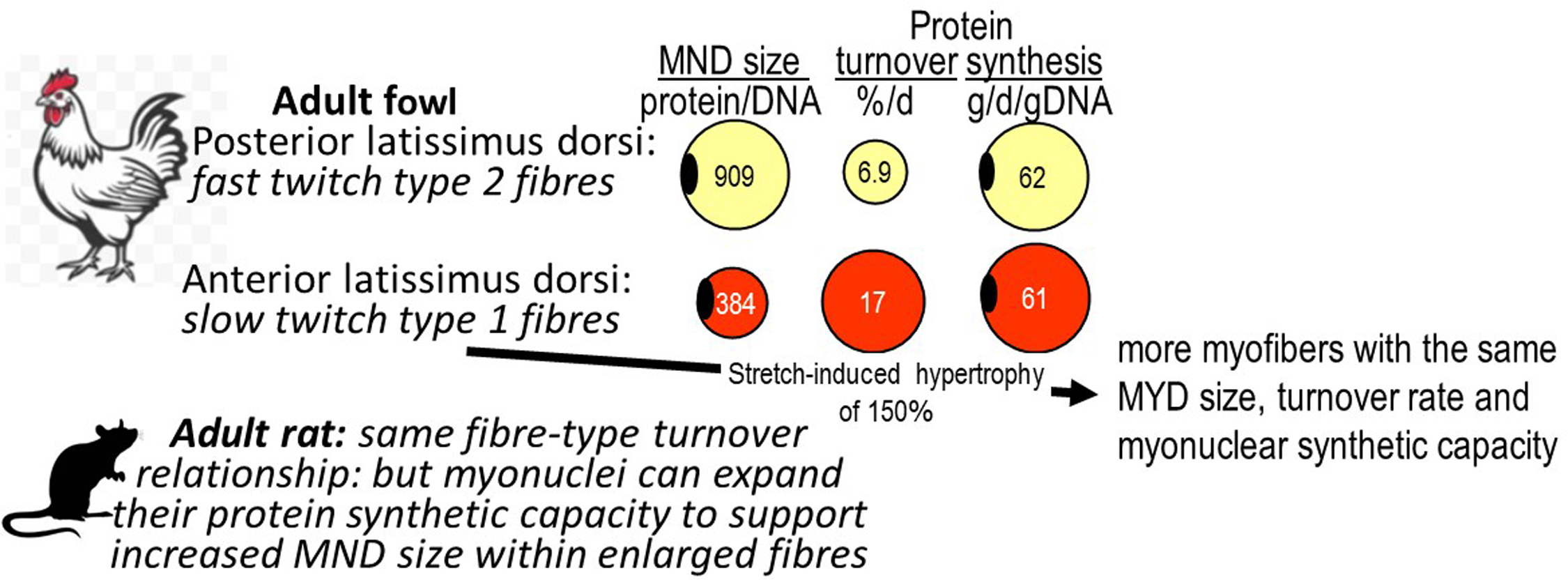
Figure 1. The myonuclear domain size and protein turnover. The myonuclear domain is the volume of myoplasm deemed to by regulated by a single myonucleus within the multinucleated myofibre syncytium, with units of the protein/DNA ratio. Data for the fowl from.(Reference Laurent, Sparrow and Bates37–Reference Laurent and Millward39) and for the rat from.(Reference Millward, Garlick and Stewart20,Reference Waterlow, Garlick and Millward66,Reference Millward, Bates and Laurent69,Reference Bates and Millward70)
As for the source of new myonuclei during muscle growth, the consensus is that they mainly derive from satellite cells, (SCs, muscle stem cells), after their activation, proliferation and fusion with the myofibre(Reference Millward9,Reference Millward45) .
Protein turnover and muscle growth
The key features of protein turnover and muscle growth in relation to proteostasis(Reference Millward8) are shown in Figure 2. The relative importance of MPS and MPD in muscle growth control was identified in early nutritional studies(Reference Millward, Garlick and Nnanyelugo71), with MPD necessary for structural remodelling during early rapid growth but with MPS the main target for regulation of protein deposition. This contrasted with the liver, (and possibly other visceral tissues), where proteolysis is the important control site for regulation of tissue protein mass(Reference Waterlow, Garlick and Millward66). Proteolysis is also most important in the regulation of whole-body protein turnover(Reference Fereday, Gibson and Cox72) most likely because a major component of this is turnover within the liver and splanchnic bed. MPS was shown to reflect translational capacity as ribosome content consequent to nucleolar ribosomal biogenesis, and expressed as the total RNA concentration (RNA/protein ratio), of which ≥ 85 % is ribosomal, and translational control determining translational efficiency, expressed as protein synthesis/unit RNA. Translational capacity and ribosomal efficiency proved to be a simple but effective way of identifying mechanisms of changes in MPS. Thus, in malnourished rats, the reduced MPS mediating growth suppression involved reductions in both translational capacity and efficiency, with the former mediating the major change during longer-term deficiency(Reference Millward, Garlick and James73). Turnover was shown to be rapid during early post-natal growth(Reference Millward, Garlick and Stewart20) or during muscle hypertrophy(Reference Laurent, Sparrow and Millward38) due to the structural remodelling of myofibers.

Figure 2. Key aspects of the regulation of proteostasis and turnover in muscle. Protein turnover is shown as part of proteostasis, an umbrella term describing all aspects of the maintenance of an intact proteome within cells, acting through a proteostasis network, (PN)(Reference Millward8) to control the initial production, (protein synthesis), folding, conformational maintenance, abundance, subcellular localisation, and disposal by breakdown mainly by the lysosomal-autophagic system and ubiquitin–proteasomal systems. Detail and references for the key findings shown are given in the text.
The role of insulin was established in early studies(Reference Millward, Odedra and Bates29,Reference Odedra, Dalal and Millward34,Reference Wool and Cavicchi74–Reference Anthony, Lang and Crozier78) emerging as the main acute, nutritionally regulated, (especially by protein), mediator of muscle protein synthesis regulation(Reference Millward26,Reference Millward27) . The interaction of amino acids with insulin and the extent of their independent influence on MPS in mediating the feeding response, initially proved difficult to establish in these rat studies. When they were both controlled independently it was demonstrated that insulin was permissive for the maximum influence of amino acids, especially leucine(Reference Anthony, Lang and Crozier78–Reference O’Connor, Kimball and Suryawan81) with insulin at low physiological levels and amino acids acting through independently upstream pathways to activate an mTORC1-mediated stimulation of MPS(Reference Millward9). In adult human skeletal muscle, work by Mike Rennie, Phil Atherton, Kenny Smith and colleagues have shown that amino acids are the main regulatory influence on protein synthesis, with insulin acting primarily to inhibit proteolysis(Reference Millward and Smith47). Dietary-protein mediated changes in thyroid hormones also emerged as important in the regulation of both muscle protein synthesis at the level of the ribosomal capacity and MPD(Reference Brown, Bates and Holliday31–Reference Brown, Van Bueren and Millward33), so that reductions in both insulin and thyroid hormones, (specifically circulating free-T3) were shown to mediate protein deficiency-induced reductions in muscle growth and protein turnover(Reference Jepson, Bates and Millward30).
Influence of dietary protein on bone and muscle growth, muscle protein and connective tissue synthesis and the role of IGF-1
The concern that protein deficiency was a major world wide problem in the 1960s(Reference Semba82) resulted in the development of animal models of protein deficiency within Platt’s MRC Human Nutrition Research Unit at Mill Hill(Reference Platt and Stewart83,Reference Platt, Heard, Stewart and Munro84) . Platt also held the Chair of Human Nutrition at the LSHTM and by the time Waterlow succeeded Platt to the Chair in 1970, several of Platt’s research group, Heard, Payne, and Stewart, were at the LSHTM and work on a rat model of marginal protein deficiency was on going(Reference Payne and Stewart85,Reference Stewart, Preece and Sheppard86) . I was able to take advantage of this work by studying muscle growth and protein turnover over a year of growth and development of these two colonies(Reference Millward, Garlick and Stewart20). The growth of male rats from these colonies is shown in Figure 3. The malnourished rats were smaller, (40 % reduction in body and muscle weight), and shorter, (12 % reduction in body and femur length): i.e. they were stunted. We showed that the impaired muscle growth reflected impaired muscle protein synthesis, especially in early life when myofibre formation would have normally been intense, with a marked delay in the usual early increase in myonuclei and with a much reduced translational activity responsible for the impaired muscle protein synthesis and growth.
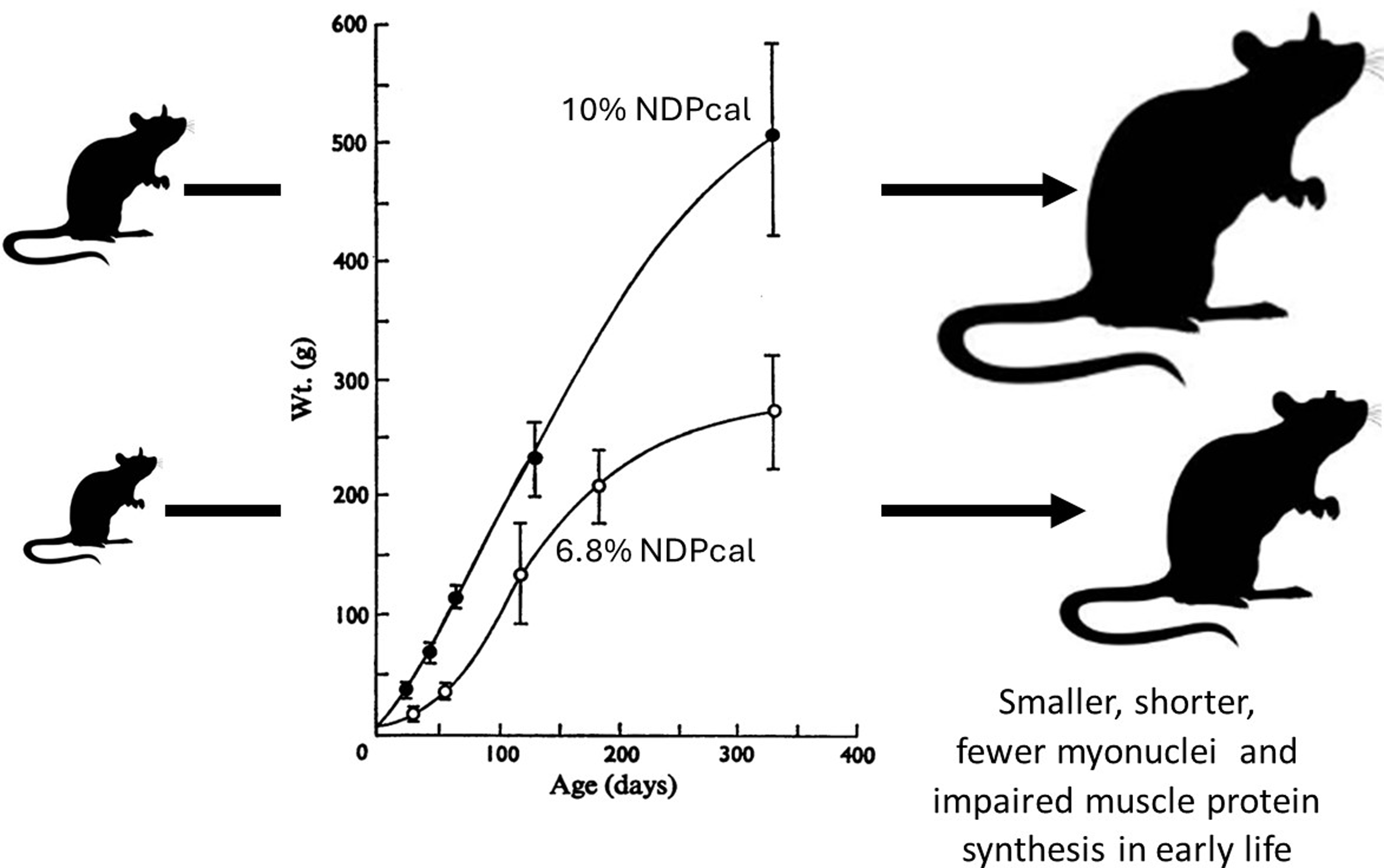
Figure 3. Influence of dietary protein on growth in well fed and multigenerational marginally malnourished rats. Data from(Reference Millward, Garlick and Stewart20,Reference Payne and Stewart85,Reference Stewart, Preece and Sheppard86)
We returned to the issue of the impact of protein deficiency on muscle and bone growth in the late 1980s when Zainal Yahya joined us to investigate the interactions between bone and muscle growth and dietary protein, specifically the role of IGF-1 since its role in post-natal muscle growth has been difficult to untangle and often controversial (see(Reference Millward9)). After demonstrating the relationship between growth in muscle mass (the gastrocnemius) and in bone length (the tibia)(Reference Yahya and Millward42), discussed above, we undertook studies related to remodelling of the extracellular matrix in muscle, which is necessary for increases in muscle cross-sectional area and length during growth. We assumed there would be an active synthesis of muscle connective tissue synthesis, (MCTS), occurring in well-fed rapidly growing rats, measurable in terms of proteoglycan synthesis, which should diminish as growth slowed with protein deficiency. If these changes were mediated by an autocrine/paracrine action of IGF-1 in muscle in response to passive stretch from bone length growth, we should observe reductions in muscle IGF-1 as growth slowed in parallel to reductions in proteoglycan synthesis (PGS), and this was the case. As shown in Figure 4(a), in response to graded protein deficiency, while bodyweight growth stopped more or less immediately with all deficient diets in association with the hypoinsulinemia, inhibition of tibial length growth was markedly delayed during which time some muscle growth occurred as the rats got longer and leaner with increasing protein deficiency and time(Reference Yahya and Millward42). Muscle IGF-1 concentrations fell(Reference Yahya, Bates and Tirapegui40,Reference Yahya, Bates and Millward41,Reference Yahya, Tirapegui and Bates43,Reference Yahya87,Reference Yahya, Bates and Tirapegui88) (Figure 4(b)), as tibial growth was gradually inhibited, as indicated by the width of the growth plate, and in parallel with reductions in MCTS, falling to very low levels as growth slowed and ceased(Reference Yahya, Tirapegui and Bates43). We observed reductions in insulin, (and plasma IGF-1, highly correlated with insulin(Reference Yahya, Bates and Tirapegui40)), and in muscle protein synthesis(Reference Yahya, Tirapegui and Bates43) which was consistent with hypoinsulinemia mediating the reduced muscle protein synthesis.
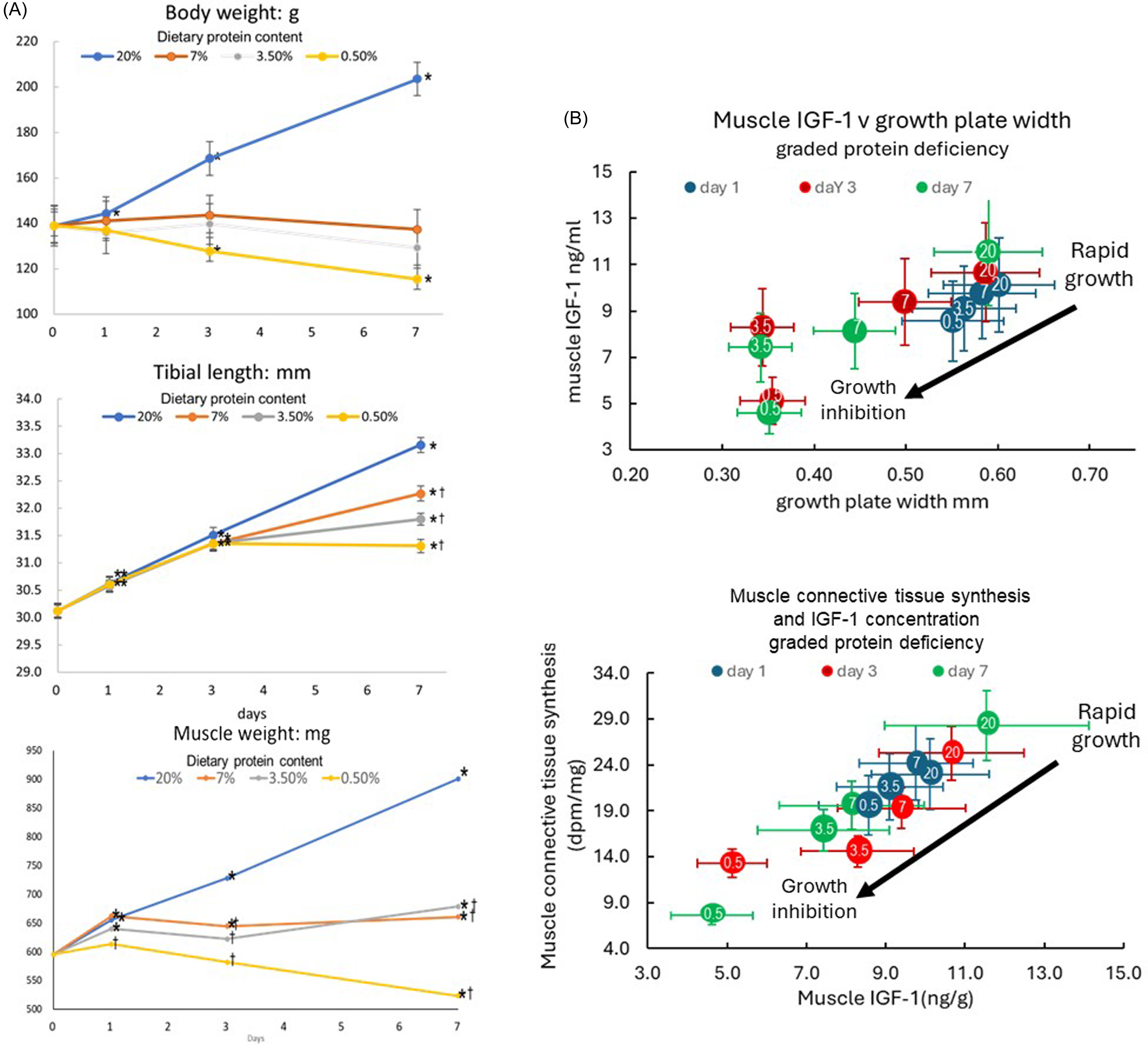
Figure 4. Responses of growth, muscle IGF-1 and connective tissue synthesis to graded protein deficiency. Young rapidly growing rats fed diets of graded protein deficiency (20 %,7 %, 3·5 % and 0·5 % protein with responses of body weight, tibial length and gastrocnemius muscle weight measured after 1, 3 and 7 d shown in panel A(Reference Yahya and Millward42), and muscle IGF-1 in relation to tibial growth (as indicated by the width of the growth plate, panel B top) and muscle connective tissue synthesis, (as indicated by 35S incorporation after a large dose of 35S sulphate, panel B bottom(Reference Yahya, Bates and Tirapegui40,Reference Yahya, Bates and Millward41,Reference Yahya, Tirapegui and Bates43,Reference Yahya87,Reference Yahya, Bates and Tirapegui88) ). In Panel B, dietary protein content is shown as the numbers within the coloured circles with the time after diet change in days indicated by the circle colour shown in the legend
Although these were only associative studies, the finding are consistent with the regulation of IGF-1 within muscle being separate from that of circulating, endocrine IGF-1 function, and occurring in response to mechanotransduction associated with bone length growth during post natal growth. Thus muscle IGF-1 was a driver of ECM remodelling(Reference Yahya, Bates and Tirapegui40–Reference Yahya, Tirapegui and Bates43,Reference Yahya87,Reference Yahya, Bates and Tirapegui88) , i.e. ‘bag enlargement’, and insulin and amino acids mediated myofibrillar protein synthesis and ‘bag filling’. As discussed elsewhere(Reference Millward9), local IGF-1 production is now known to be part of the mechanotransduction of stem cell activation and myogenesis.
The current model for regulation of muscle and bone growth
The growth model consistent with the current evidence base is shown in Figure 5 and has recently been discussed in detail(Reference Millward9,Reference Millward45) .
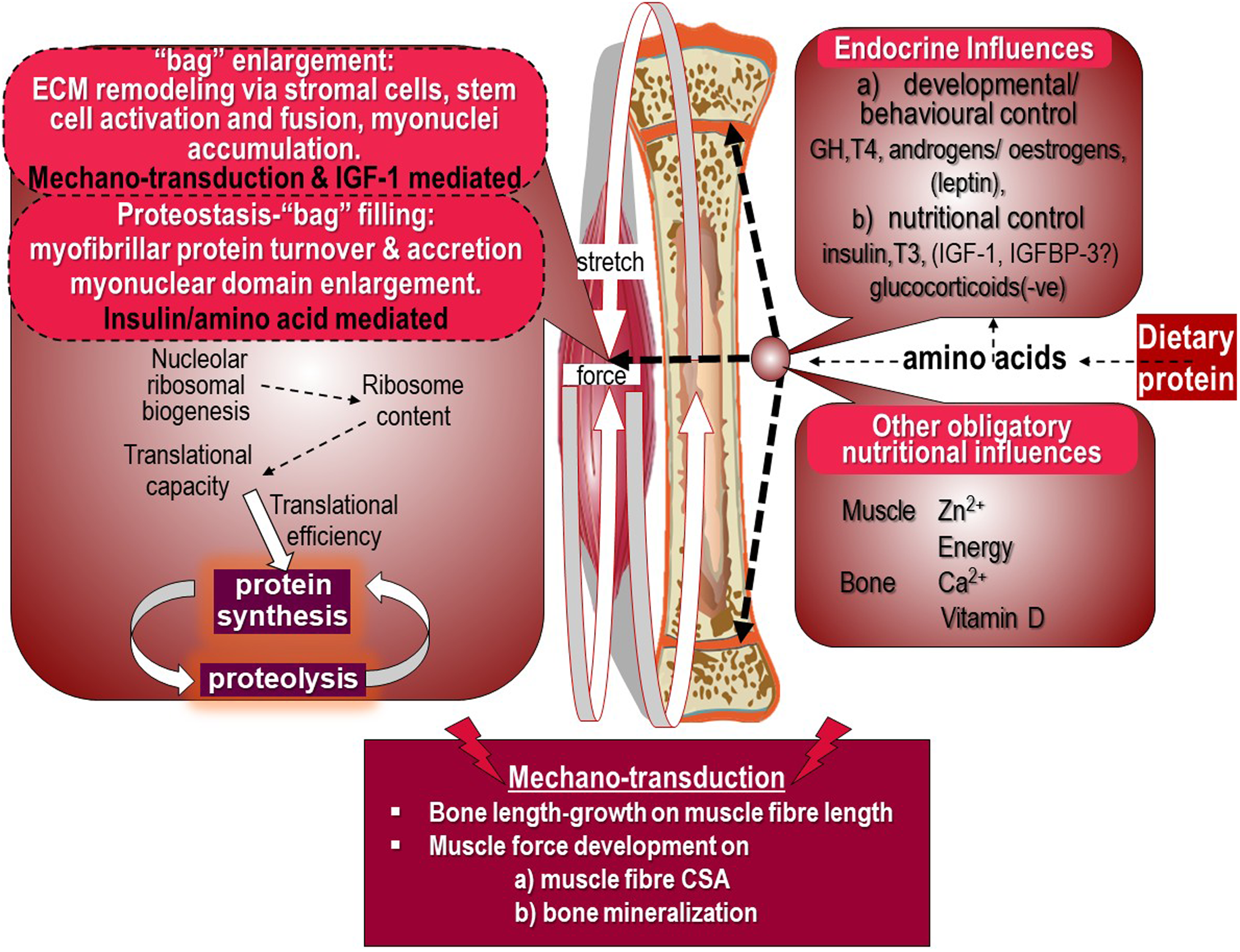
Figure 5. Dietary protein and appendicular muscle-bone interactions in the rat. The control of the growth of the major appendicular muscles is directly related to the lengthening of their associated bone through endochondral ossification in the growth plate which occurs in response to dietary protein, endocrine responses and other obligatory nutrients which also act on muscle to induce growth. The mechanotransduction of muscle length growth reflects passive stretch from bone lengthening which mediates addition of sarcomeres. Force development through muscle contractile activity in the lengthening muscle in response to gravitational loading induces growth in muscle cross-sectional area and also acts on osteocytes in bone to promote mineralisation to increase bone strength. Muscle growth is shown as bag enlargement, part mediated by mechanotransduction and the autocrine/paracrine action of IGF-1, and bag filling mediated by amino acids and insulin. Details are reviewed in(Reference Millward9)
The growth of an appendicular muscle reflects the combined influence of nutrition, especially amino acids from dietary protein driving both muscle and bone length growth, and mechanotransduction exerted both by stretching, consequent to length growth of the associated bone, and by forces generated by internal work against gravity. Other key nutrients are obligatory for muscle growth of which zinc is most important(Reference Giugliano and Millward22,Reference Giugliano and Millward23,Reference Millward55) , in part to enable dietary protein’s anabolic influence and also because zinc-dependent enzymes are widespread in muscle. Calcium and vitamin D are also important for bone growth and strength.
Dietary protein is shown to control both muscle and bone growth through the combined effects of amino acids and the endocrine responses which are under behavioural/developmental and nutritional control. In the male rat, muscle and length growth continues throughout its lifespan, since epiphyseal growth plate fusion does not occur. The multiple targets of dietary protein’s influence include endochondral ossification in the bone growth plate mediating bone length growth, and myonuclear domain enlargement through myofibre protein deposition.
Muscle growth is identified in the left panel of Figure 5 under two headings. The first is ‘bag’ enlargement, which includes ECM remodelling via stromal cells, stem cell activation, proliferation and fusion and myonuclei accumulation within the myofibre allowing myofibre expansion in length and width, under control by mechanotransduction within which IGF-1 links such forces to anabolic processes. The second, under the heading of proteostasis which involves ‘bag’ filling, which is myofibrillar protein turnover and accretion resulting in myonuclear domain enlargement under the control of insulin and amino acids. MPS is shown to reflect both translational capacity as ribosome content consequent to nucleolar ribosomal biogenesis, and translational control determining translational efficiency.
The mechanotransduction shown in Figure 5 involves bone length-growth acting to mediate muscle fibre length through passive stretching and muscle force development during work against gravity. Which acts to increase both muscle fibre CSA(Reference Millward9), and bone mineralisation(Reference Frost89). The targets of these mechanical forces in muscle are activation of satellite cells (SCs) and myogenesis, and remodelling of the extracellular matrix which increases the capacity for myofibrillar protein accretion within the myofibre syncytium.
Key implications of this growth model for childhood growth have been discussed elsewhere(Reference Millward45). During the pubertal growth spurt if this growth model is correct, the time course of changes in bone and muscle growth in puberty should be that bone length-growth peak velocity precedes muscle CSA peak velocity which precedes full bone mineralisation, and this has been observed. During a seven-year longitudinal study of lower leg growth during puberty in Finnish girls(Reference Xu, Nicholson and Wang90), peak growth velocities (PGV) measured with respect to menarche occurred first for tibial length and for total bone CSA at 20 months prior to menarche, with PGV for muscle CSA 1 year later and with the final PGV, for cortical volumetric bone mineral content, occurring 8 months later at menarche. While tibial length growth was complete at 18 years of age all other growth measures were incomplete, continuing into early adult life when full maturation is achieved.
Maintenance
Apart from early infancy and the pubertal growth spurt, human growth is slow and probably saltatory, (i.e. episodic)(Reference Lampl, Veldhuis and Johnson91–Reference Lampl and Johnson93), so that humans are mainly at maintenance throughout their post infancy life and controlling the maintenance of the muscle and the FFM must be a major component of any general control mechanism. As I have recently reported(Reference Millward94), the literature suggests that the phenotypic skeletal muscle mass in well-fed adults with healthy lifestyles and not engaging in specific resistance exercise does not vary as a function of their protein intake within a wide range of intakes. This seemed obvious to us in the 1980s and we showed it to be the case in underwater weighing studies of body composition in various groups of adult men and women i.e. athletes (elite rowers and natural body builders), the obese and healthy men and women. While FFM was highest in the male athletes and lowest in normal healthy females, in each group FFM varied with height with the same slope and the higher levels in the athletes reflected their phenotype and training(Reference Millward44). Whilst there was no obvious influence of dietary protein intake, homeostatic regulation was likely to occur in response to variation in habitual protein intake and we set out to investigate this.
Our early stable isotope studies based on GC-MS techniques facilitated human studies of the response of whole body and muscle protein turnover(Reference Rennie, Edwards and Halliday46) to feeding, but were largely exploratory demonstrating that protein turnover in humans in vivo could be measured and was responsive to protein intake. Subsequently, we undertook more focussed studies to investigate the response of whole-body protein turnover and homeostasis in response to varying protein intake in two series of studies.
Adaptive diurnal cycling
We first studied the diurnal changes in both N balance and amino acid balance and protein turnover with multiple stable-isotope tracers at the end of 2 week periods of adaptation to a range of protein intakes with an 8 h continuous iv tracer fasting-feeding protocol. We found, with both 12 h nitrogen (N)-balances, corrected for changes in size of the body urea pool, and [13C-1] leucine balances, a nutritionally sensitive diurnal cycle of increasing fasting protein and N losses and fed state gains of increasing amplitude with increasing protein intakes(Reference Price, Halliday and Pacy48,Reference Pacy, Price and Halliday49) . The same protocol was also used to study the time course of the adaptation of the diurnal cycle of N homoeostasis over 9–14 d during a change in protein intake from a high to a moderate protein intake(Reference Quevedo, Price and Halliday50). This showed that adaptation of amino acid oxidation to a change in protein intake was slow and incomplete over the short time period of our studies and involved some transient weight loss while the adaptation was occurring. These results were the basis for the maintenance model showed in Figure 6. In effect, homeostasis during the diurnal cycle involves bag emptying and refilling within muscle with a regulated level of the FFM set primarily by a skeletal muscle ‘bag full’ mechanism as demonstrated with studies of postprandial muscle protein synthesis in adults(Reference Bohé, Aili Low and Wolfe95–Reference Mitchell, Phillips and Hill98). The regulated whole-body protein level does include a minor component likely to increase with habitual protein intake, i.e. the splanchnic protein mass which is not limited. This has been demonstrated in men fed increased protein intakes for 10 weeks who showed increased trunk protein but no change in appendicular lean mass(Reference Mitchell, Milan and Mitchell99), consistent with animal studies.
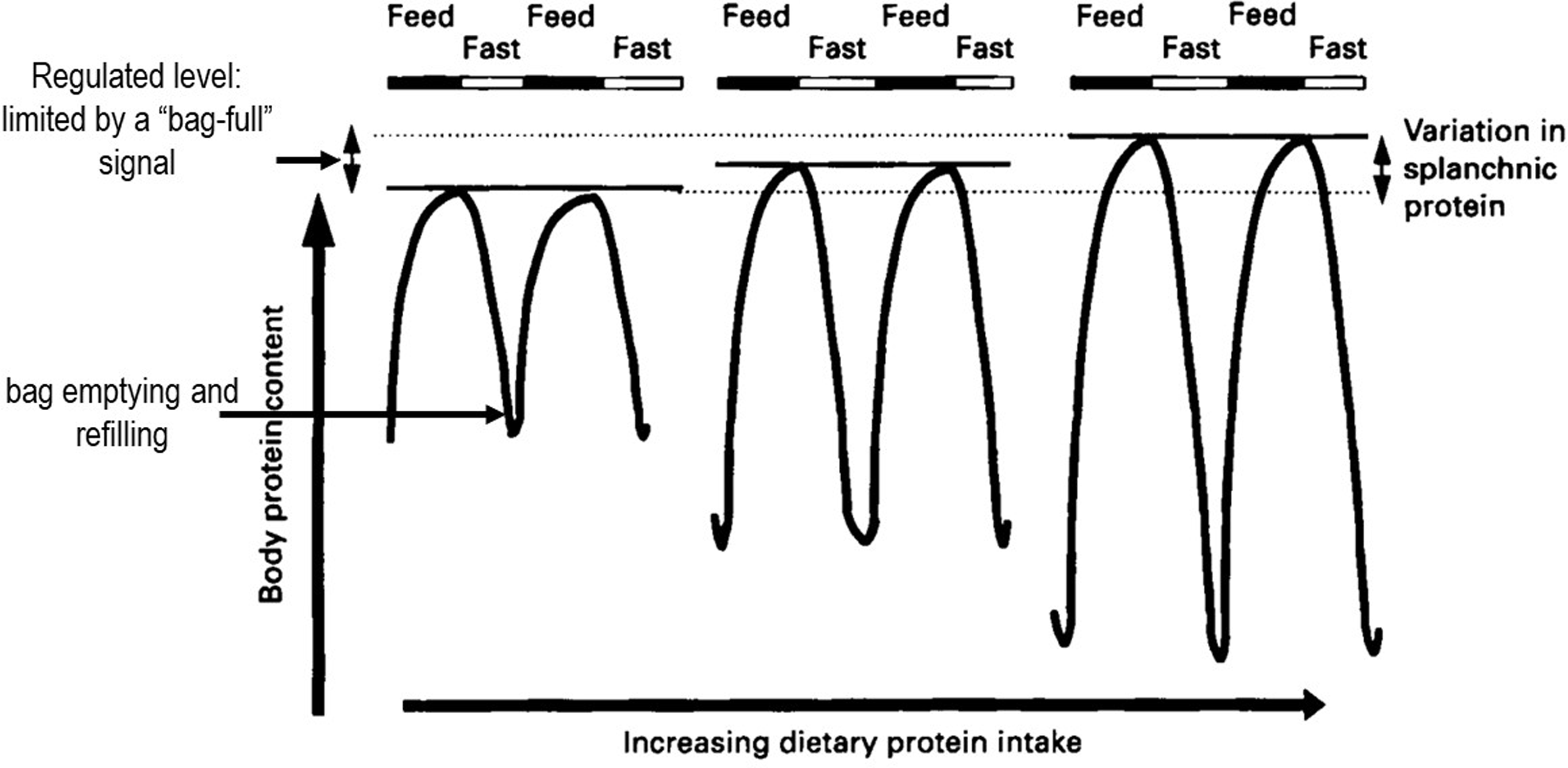
Figure 6. Diurnal cycling of body protein and adaptive changes in response to increasing habitual protein intake. There is a nutritionally sensitive diurnal cycle of fasting N and protein losses and fed state gains of the fat-free mass, of increasing amplitude with increasing habitual dietary protein intakes, mainly reflecting ‘bag’ emptying and refilling in skeletal muscle. Model-based on data from multiple stable isotope studies both in adults fed various levels of dietary protein for 2 weeks(Reference Price, Halliday and Pacy48,Reference Pacy, Price and Halliday49) and after the change in intake from a generous protein level to that of the current RDA(Reference Quevedo, Price and Halliday50). The regulated level of the FFM, set by the skeletal muscle ‘bag full’ level mechanism(Reference Bohé, Aili Low and Wolfe95–Reference Mitchell, Phillips and Hill98), does include a minor component known to increase with habitual protein intake which involves the splanchnic protein mass which is not specifically limited.
Postprandial protein utilisation
The second series of studies were designed to study the regulation of postprandial protein utilisation(Reference Gibson, Fereday and Cox52) and to quantify its efficiency,(Reference Fereday, Gibson and Cox51,Reference Fereday, Gibson and Cox72,Reference Millward, Fereday and Gibson100–Reference Millward, Fereday and Gibson102) . The efficiency of postprandial protein utilisation, (PPU, the equivalent of the classical term NPU, net protein utilisation), was important since it had been suggested in the 1985 FAO/WHO/UNU protein and energy requirements report that the elderly needed more protein because of an age-related decline in protein utilisation, although no unequivocal evidence had been cited to support this. Also it was noted in the 2007 report(103) that the efficiency of protein utilisation in healthy adults consuming high quality dietary sources, as indicated in N-balance studies, appeared from the slope of the balance data to be usually very low, often ≤ 50 %, which was biologically implausible. Our 9 h protocol(Reference Gibson, Fereday and Cox52), a primed continuous infusion of [13C-1] leucine involving 3 × 3 h periods, (post absorptive then isoenergetic feeding of low and then habitual protein in small meals), measuring leucine oxidation and balance, enabled PPU to be calculated as ∆balance/∆intake from the two feeding phases. This indicated a combination of an insulin-mediated, protein-conserving influence of dietary energy that inhibited whole body protein degradation, lowered amino acid levels, and reduced oxidation, and an amino acid-mediated augmentation of the inhibition of degradation, a stimulation of protein synthesis, and an increase in oxidation when leucine dietary supply exceeds the capacity for its net deposition(Reference Gibson, Fereday and Cox52,Reference Fereday, Gibson and Cox72) . The efficiency of protein utilisation, the PPUnitrogen value, (calculated from the leucine balance data adjusted for the difference in the leucine/N ratios of protein in food compered with tissues), was what would be expected, high for milk, close to unity (100 %), and lower for wheat(Reference Millward, Fereday and Gibson100), (see Table 1). Furthermore we were able to show that the efficiency of protein utilisation in individuals, and a component of their apparent protein requirement, is determined by the sensitivity of the insulin-mediated inhibition of whole body proteolysis to amino acid supply(Reference Fereday, Gibson and Cox72).
Table 1. Protein intakes and apparent requirements in healthy adults*
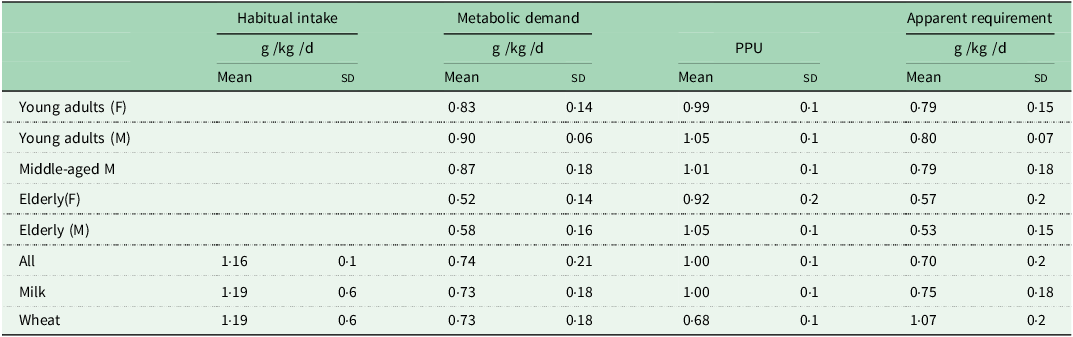
* Values from Millward 2004(Reference Millward54).
The Adaptive metabolic demand model for homeostatic regulation of the fat-free mass
The results of these two sets of studies of diurnal cycling and postprandial protein utilisation enabled us to formulate a new model for the protein requirement based on the concept of an adaptive metabolic demand(Reference Millward54) and this allowed us to add more detail to the homeostatic regulation of the fat-free mass throughout the diurnal cycle of feeding and fasting as shown in Figure 7. In this model, the dietary protein requirement, (metabolic demand/efficiency of utilisation) involves a metabolic demand for amino acids for maintenance which comprises two components. The first is small and fixed, which is the sum of those processes which irreversibly consume amino acids for various purposes and which contribute to the obligatory nitrogen losses, (about 0·3 g protein/kg/d)(Reference Rand, Pellett and Young104). The second is a variable adaptive component driven by oxidative losses of the essential amino acids (EAAs). One possible metabolic explanation of this relates to the homeostatic mechanisms which maintain the low tissue concentrations of the potentially toxic branched chain, aromatic and S-containing amino acids(Reference Rennie, Edwards and Krywawych105,Reference Bergström, Fürst and Norée106) . Thus during the usual relatively slow growth in childhood, and at weight maintenance in adults, the supply of these amino acids from food protein will usually be markedly in excess of minimal needs so that they must be rapidly disposed to avoid excessive postprandial increases in their tissue concentrations. This requires that the capacity, (Vmax), of the pathways of oxidative catabolism of these particular amino acids adapts to match the habitual protein intakes(Reference Millward107,Reference Millward and Rivers108) . Although these pathways are to some extent regulated by feeding and fasting, this regulation is only partial, so that amino acid oxidation continues to occur after dietary protein is disposed of, continuing in the postabsorptive state with net catabolism of tissue protein. This means that in practice, adaptive oxidation is relatively insensitive to acute food or protein intake, but does change slowly with a sustained change in intake, enabling N equilibrium to be achieved eventually within the range of intakes to which adaptation can occur. The lower limit of this, which is yet to be identified, determines the minimum protein requirement.
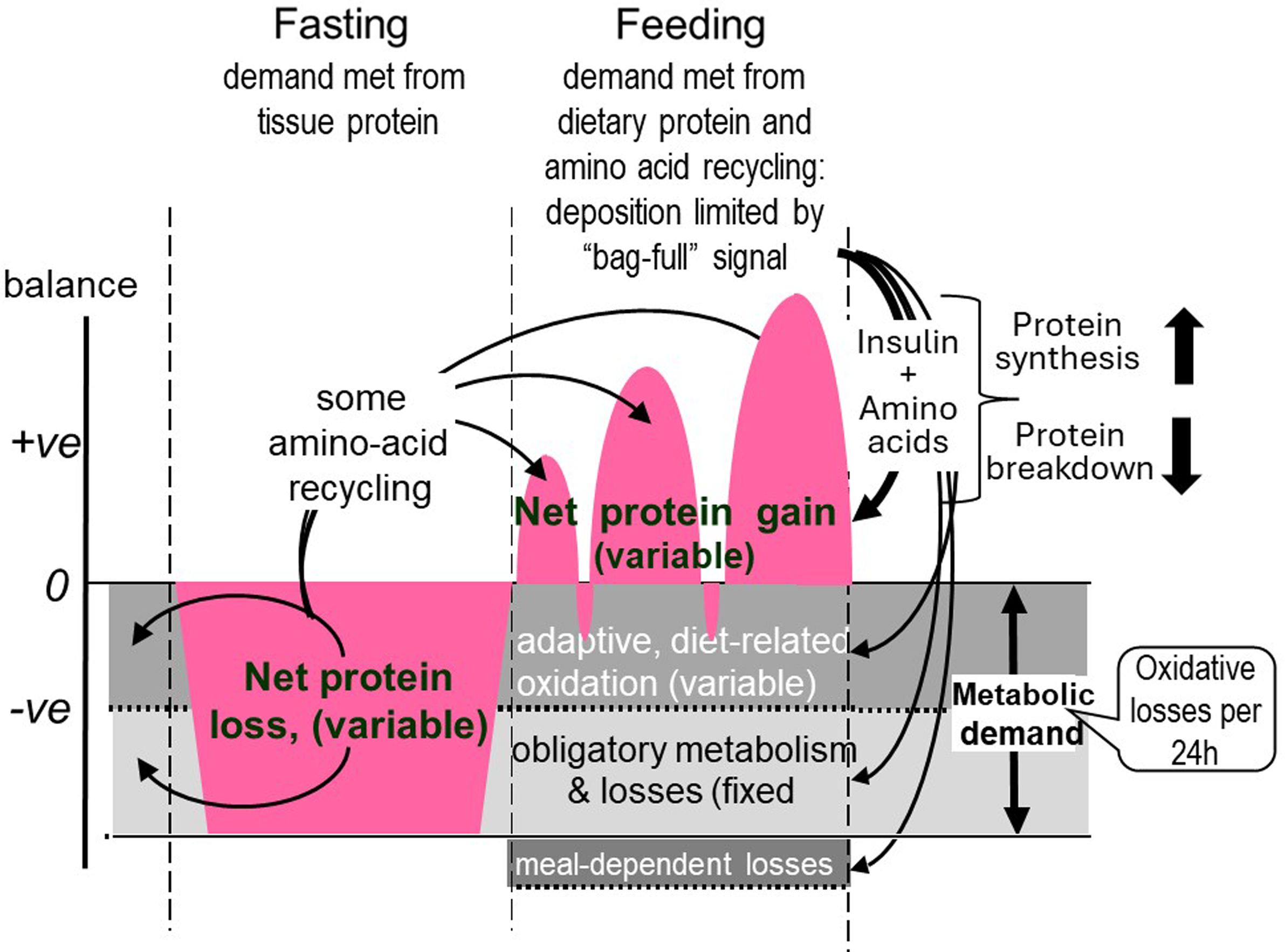
Figure 7. Adaptive metabolic demands throughout the diurnal cycle. The metabolic demand for dietary protein and amino acids, comprise two components, an obligatory fixed demand for amino acids as precursors for various metabolites which are eventually degraded and excreted as CO2 and the obligatory nitrogen losses, and an adaptive demand which is amino acid oxidation set at a rate which varies with habitual protein intakes. These demands are assumed to occur at a constant rate throughout the 24 h cycle and change only slowly with a change in habitual intake so that fasting oxidative losses and nitrogen excretion scaled to 24 h indicates a first approximation of the metabolic demand. During fasting the demand is met from tissue protein and with feeding dietary protein provides for the demand and for repletion of fasting losses with some recycling of some essential amino acids after their release during fasting. The IAA content of the metabolic demand is variable according to the magnitude of the demand so that protein quality is difficult to predict.(Reference Millward and Pacy101,Reference Millward109)
In the fasted state tissue protein is mobilised to provide amino acids for both the fixed, obligatory and variable (adaptive) components of the metabolic demand. With feeding, dietary amino acids are utilised to replace tissue protein mobilised in the post absorptive state and to provide for the obligatory and adaptive metabolic demands. As previously demonstrated(Reference Millward, Fereday and Gibson100) and discussed(Reference Millward and Pacy101,Reference Millward109) for some amino acids, recycling may occur, namely those liberated in the fasted state which are not completely oxidised. However, the extent of this is difficult to quantify but will vary according to the kinetics and sensitivity to changes in pool sizes of the catabolic pathways of each amino acid, but is more likely with lysine(Reference Millward, Fereday and Gibson100) and threonine. Recycling may also vary with the habitual level of protein intakes which governs the amplitude of fed state gains and fasting losses of tissue proteins(Reference Millward109). The minimum protein requirement would be the sum of the fixed obligatory demand and the lower bounds of the adaptive demand. The amino acid pattern of the demand which is for both protein deposition and for the obligatory and adaptive components of oxidative losses, is difficult to predict(Reference Millward and Pacy101,Reference Millward109) . The demand for protein deposition requires a pattern of amino acids like that of tissue protein although any recycling as shown in Figure 7 will to some extent reduce the dietary demand for some amino acids. Subjects with higher habitual protein intakes will display an increased amplitude of tissue gains and losses as in Figure 6 and although the extent of recycling may be increased, the EAA requirement pattern is likely to increasingly reflect that of tissue protein as with rapidly growing animals when the maintenance component will be a proportionally smaller component.
Any excess amino acid intake is oxidised (shown as meal-dependent losses), and it is the extent of this component that determines the efficiency of dietary protein utilisation. Thus more realistic efficiency values can be measured within an experimental framework that takes account of the adaptive metabolic demand. In contrast to the current requirements model, for fully adapted individuals, risk of deficiency, (i.e. negative N balance after complete adaptation), will only start to increase when habitual intakes fall below the range of the true minimum requirement. This value is currently unknown, but may be at the lower end of the reported distribution of requirements as indicated by N balance studies, i.e. between 0·4 and 0·5 g/kg per d(Reference Rand, Pellett and Young104), or at intakes at the lower end of the Acceptable Macro-nutrient Distribution Range (AMDR), for protein: 10–35 % of daily calorie intakes(110)). At intakes greater than this, within the AMDR, the additional metabolic demands vary directly with the intake so that deficiency is only likely as a short-term response to a change to a lower intake within the adaptive range. In practice, few natural diets which provide requirement levels of all other nutrients, provide protein intakes as low as this(Reference Millward54). On the basis of measured metabolic demands (MD), post absorptive losses scaled to 24 h in subjects on their habitual protein intakes or after 2 weeks adaptation, and PPU (fractional efficiency of protein utilisation) calculated from leucine balance converted to nitrogen balance, an apparent protein requirement can be calculated as MD/PPU. As shown in Table 1 for young and middle-aged men and women on habitual diets estimated at 1·16 ± 0·08 g/kg/d apparent requirements were 0·79–0·80 g/kg/d falling to somewhat lower values in the elderly because of significantly lower values for the MD(Reference Fereday, Gibson and Cox51,Reference Millward, Fereday and Gibson53) and in adult men and women on similar habitual protein intakes fed either milk or wheat protein-based small meals(Reference Millward, Fereday and Gibson100) apparent requirements were 0·75 and 1·07 g/kg/d for milk or wheat protein meals, the higher value for wheat because of the lower PPU value due to lysine limitation. We could also calculate the lysine requirement from the milk and wheat protein studies and the value, 23·2 ± 2·0 mg/kg/d was somewhat lower than other values reported from stable isotope studies but closer to earlier reports from nitrogen balance studies(103).
Whilst this adaptive metabolic demand model for protein requirements has not been widely adopted no alternative model has been proposed which could account for all the observed characteristics of postprandial metabolism and nitrogen balance studies. Although higher values have been reported for adults, (and for children), determined by the Indicator Amino Acid Oxidation method, a recent critical analysis of most of the results obtained by this method shows that the experimental design is critically flawed by the limiting amounts of the indicator amino acid fed in the test meals in the studies(Reference Millward94).
Food intake regulation
As illustrated in Figure 7 the metabolic demand for protein involves repletion of post absorptive protein losses, any growth, and the obligatory and adaptive components of oxidative amino acid losses. Understanding how food protein intake is adjusted to match these demands is challenging. The original protein stat model included an aminostatic mechanism(Reference Millward44,Reference Melinkoff, Frankland and Boyle111) , sensing the relative amino acid balance between the circulating amino acid pool and uptake into tissues, activating appetite to mediate feeding and inhibiting it when amino acid uptake into muscle became limiting by a bag-full’ signal. This was in line with Webster’s view, based mainly on animal studies, that the demands for protein generated by LBM growth was the main driver of food intake(Reference Webster112). Food intake measurements in rats fed marginally protein-deficient diets by us and others(Reference Jepson, Bates and Millward30,Reference Yahya and Millward42,Reference Coyer, Rivers and Millward113,Reference Laeger, Reed and Henagan114) , had shown hyperphagia consistent with Websters idea and this sort of data became the basis for Gosby & Simpsons protein leverage hypothesis(Reference Gosby, Conigrave and Raubenheimer115). Also our rat studies of the response to severe zinc deficiency had shown the characteristic cycles of anorexia-induced wasting and refeeding associated with cyclic changes in zinc availability as it was sequestered and then released from muscle(Reference Giugliano and Millward22,Reference Giugliano and Millward23) , a response abolished by a low protein diet(Reference O’Dell, Reeves and Mills116), and reported at the time to be mediated by satiating intakes of the indispensable amino acids (methionine, phenylalanine, threonine and tryptophan)(Reference Chesters and Will117)
Human studies of appetite regulation usually involve subjects at maintenance and these show that the size of the FFM drives food intake(Reference Blundell, Caudwell and Gibbons118,Reference Blundell, Gibbons and Beaulieu119) , although the mechanistic link is seen as energy expenditure. However, Dulloo and colleagues have discussed both passive and active roles for the FFM in appetite regulation(Reference Dulloo, Jacquet and Miles-Chan120). The passive role, mediated by ‘energy-sensing’ mechanisms that translate FFM-induced energy requirements to energy intake should operate most of the time, with the active role limited to dynamic changes in the FFM in body weight recovery during refeeding, when a relationship between deficits in FFM and increased food intake occur(Reference Dulloo, Jacquet and Girardier121). Catch up growth in malnourished children is an example of an active role of the FFM, especially of muscle mass, in the activation and inhibition of appetite which could involve an aminostatic appetite mechanism, although restoration of the body fat mass is also likely to play a role(Reference Friedman122). Studies in low birthweight infants showed that tolerance of increasing dietary protein intakes was at its limit at maximal rates of protein accretion(Reference Kashyap, Schulze and Forsyth123). Growth in normal infants and prepubertal children has been reported to involve coupled saltatory growth in length(Reference Lampl, Veldhuis and Johnson91) and head circumference(Reference Lampl and Johnson93) with 90–95 % of infant development growth-free. Thus infant growth is said to occur by intense occasional bursts, and with longer less intense bursts of length growth in prepubertal children(Reference Tillmann, Thalange and Foster124). The precise relationship between these length growth spurts and food intake is not known with any precision because of the difficulty of making accurate daily measurements of food intake(Reference Hackett, Rugg-Gunn and Appleton125). More recently the potential role of fat-free mass in appetite control has become more widely discussed particularly in terms of the regulatory involvement of FFM in appetite-related measures with evidence suggesting that the appetite-stimulating hormone, ghrelin, has an inverse relationship with FFM(Reference Graybeal, Willis and Morales-Marroquin126).
Within the Protein-Stat,(Reference Millward44) the dietary adjustment of protein intake according to the metabolic demand for amino acids, was proposed to involve a centrally located ‘aminostatic’ amino-acid sensing mechanism(Reference Melinkoff, Frankland and Boyle111). Harpers work had shown the ability of rats to regulate intake to avoid either protein deficiency or excess but had found only a weak association between protein intake and brain serotonin levels(Reference Harper and Peters127). Gietzen had examined neural mechanisms in the responses to amino acid deficiency(Reference Gietzen128,Reference Gietzen129) , and suggested that recognition of deficiency was sensed in the anterior piriform cortex in response to a decline in the concentration of an essential amino acid which preceded adaptive changes in taste aversion.
At the time of my move to Surrey, whilst the literature generally supported dietary protein as the most satiating nutrient, the issue was not entirely settled in that some experimental designs that test the satiating effect of both the type of macronutrient and the overall energy level failed to show any difference between protein, carbohydrate and fat(Reference De Graaf, Hulshof and Weststrate130). However at Surrey, I found expertise in Linda Morgan’s group of assessing hunger and satiety in humans as well as in relation to the physiological role of the insulinotropic hormones GLP-1 and GIP(Reference Elliott, Morgan and Tredger131), and we conducted a number of investigations of appetite responses relevant to an aminostatic appetite control mechanism.
Firstly given the prediction that appetite should be suppressed when amino acid intake exceeded the demand we postulated that the satiating effect of dietary protein should fall with increasing habitual protein intakes which would increase the adaptive oxidative loss component of the metabolic demand (see Figure 7), and would be predicted to reduce postprandial amino-acidemia in response to protein intake. This was confirmed in terms of a decreased satiety in response to a high protein meal in subjects habituated to a high (1·96 g/kg/d) compared with a low (0·75 g/kg/d) protein diet(Reference Long, Jeffcoat and Millward132). Secondly, we predicted that ‘fast’ proteins which were rapidly digested and absorbed like whey(Reference Boirie, Dangin and Gachon133) producing a greater postprandial amino-acidemia would be more satiating than ‘slow’ proteins like casein which clot in the stomach, and were more slowly digested and absorbed, and this was observed(Reference Hall, Millward and Long134), (and incidentally became my most cited paper, n 1000). The whey preload was more satiating, reducing food intake from a buffet-type meal, and was associated with higher postprandial circulating levels of both amino acids and the anorexigenic gut peptides CCK, GIP and GLP-1, compared with an isoenergetic casein preload. Finally, Wendy Hall investigated the physiological mechanisms behind reports of aspartame-induced satiety and concluded that it was unlikely that aspartame increases satiety via CCK- or GLP-1-mediated mechanisms, but that small changes in circulating phenylalanine concentrations could influence appetite(Reference Hall, Millward and Rogers135). In a detailed review on aminostatic appetite mechanisms we wrote at the time, (which was not published because of hostile reviewers), while we identified a number of potential mechanisms we concluded that ‘it remains difficult to identify a coherent aminostatic mechanism of appetite regulation’.
Although these conclusions appear superficially consistent with a subsequent 2016 report that in healthy men of BMI from 22–28, protein-induced satiety, responses of appetite-regulating hormones and ad libitum energy intake were not related to a distinctive AA profile ‘contradicting the aminostatic theory’(Reference Korompokis, Östman and Dougkas136), further examination of this study is in fact quite revealing. The results show that for the branched chain amino acids (BCAs), the aromatic amino acids and methionine and tryptophan, responses of plasma concentrations to the preloads varied directly with the protein intake from little or no responses with the LP (P:E 0·089) preload to a robust increase with the HP preload (P:E = 0·40) with an intermediate response to the moderate protein preload (P:E = 0·244) with the responses still apparent after 210 min when the ad lib energy intake was measured. These ad lib intakes showed that for the BCAs, the post meal blood concentrations responses were inversely related to the energy intake. The postprandial excursions of the BCAs were as we had described in 1974 in meal-fed rats, namely a 30 % increase after a 20 % protein meal and a 50 % fall after a protein-free meal(Reference Millward, Nnanyelugo and James137).
Since our 1995 Protein-Stat paper a considerable amount of work has been done to identify protein and amino acid sensing mechanisms some of it summarised in 2012 by Fromentin & colleagues(Reference Fromentin, Darcel and Chaumontet138) who reported that amino acid consumption was detected by multiple and redundant mechanisms originating during digestion and subsequent metabolism, and recorded both directly by the central nervous system, and indirectly by vagus-mediated mechanisms.
Satiety and food aversion
Leucine, with its seven codons, is the most abundant amino acid in proteins(Reference Millward139), but its free pool size like that of the aromatic and sulphur amino acids, is regulated to maintain very low concentrations in blood and tissues(Reference Rennie, Edwards and Krywawych105,Reference Millward, Nnanyelugo and James137,Reference Millward, Davies and Halliday140) . As a result, for leucine, the ratio of its relative concentration in protein to that in free amino acid pools is very large, in rat muscle second only to phenylalanine(Reference Waterlow, Garlick and Millward66), so that postprandial excursions in leucine, (and that of some other indispensable amino acids in plasma), can vary markedly according to the balance between the dietary supply and the metabolic demand for protein. A simple conceptualisation of how hunger or satiety might be mediated by changes in the composition of the free amino acid pool in terms of the ratio of the indispensable amino acids, IAAs (especially leucine), to the DAAs (dispensable amino acids), to changes in dietary protein intake relative to the metabolic demand for amino acids, is shown in Figure 8.

Figure 8. Changes in the free amino acid pool mediating hunger and satiety. Leucine and several other indispensable amino acids have much lower concentrations in free amino acid pools compared with many dispensable amino acids but have high concentrations in protein, so that the ratio of leucine and the IAA to DAAs changes in response to the metabolic demand in relation to supply. A high demand relative to supply is predicted to lower the ratio and mediate hunger and a low demand relative to supply will increase the ratio and mediate satiety.
In fact the brain is an important component of postprandial peripheral BCA metabolism with their transamination generating neurotransmitters which are both excitatory, e.g. glutamate, and inhibitory, e.g. γ-amino butyric acid (GABA)(Reference Hutson, Sweatt and Lanoue141), as well as influencing the inward transport of tyrosine and tryptophan, (carried by the same large neutral amino acid transporter), which form norepinephrine and serotonin, respectively(Reference Fernstrom142). Because the outward transport across the blood-brain barrier is Na+ linked, intracellular concentrations of all naturally occurring amino acids in the extracellular fluid of the brain, with the exception of glutamine, are maintained at 10 % or less than the plasma concentrations(Reference Hawkins, O’Kane and Simpson143).
In 2006 central administration of leucine in the rat was shown to increase hypothalamic mTOR signalling in the paraventricular (PVN) and arcuate nuclei (ARH) of the hypothalamus and decrease food intake(Reference Cota, Proulx and Blake-Smith144). Since then, considerable advances have been made understanding leucine’s role in mediating satiety. In 2009 Blouet et al. reported that dietary leucine did increase cerebro-spinal fluid leucine concentration, which in the fasted state was 4 % of that in plasma, and which increased by 24 % after a high protein meal and by 44 % after a leucine-enriched chow meal(Reference Blouet, Jo and Li145). Furthermore, neurons in the mediobasal hypothalamus sensed the changes in leucine concentrations and reduced food intake by activation of the amino acid-sensing p70 S6 kinase 1 (S6K1) signalling pathway. Subsequently, working with primary cultures of post-weaning murine mediobasal hypothalamic neurons, and with hypothalamic neurons derived from human induced pluripotent stem cells, Blouet’s group identified both a stimulation of anorexigenic POMC neurones, with leucine activating a voltage-gated calcium channel, and a leucine inhibitory action on orexigenic NPY/AGRP neurons with inhibition of a store-operated calcium current(Reference Heeley, Kirwan and Darwish146). This was followed by in vivo studies identifying leucine sensing circuits in the brain. Firstly, hunger-suppressing leucine sensing was shown to occur in the nucleus of the solitary tract (NTS) in the brainstem, activating a neural circuit via the dorsomedial hypothalamus (DMH) to inhibit agouti-related protein, (hunger-driving), neurons in the ARH(Reference Tsang, Nuzzaci and Darwish147). Subsequently, a hypothalamic satiety-promoting response was shown to be mediated by leucine via a specific calcium channel protein Cav3·1, that is enriched in hypothalamic POMC leucine-sensing neurons(Reference Tsang, Heeley and Alcaino148). Leucine was shown to bind to a hydrophobic pocket in the protein increasing the voltage sensitivity for calcium transport into the cell and activating mTORC1 signalling which results in anorexia. GLP-1, which, as indicated above is increased in response to protein preloads as shown in our whey v. casein satiety studies(Reference Boirie, Dangin and Gachon133), also acts via its receptor on POMC neurons of the arcuate nucleus which involves another calcium channel protein TRPC5 involved in the mediation of leptin, insulin, serotonin, and GLP-1 satiety signalling via POMC neurons. However, according to Tsang et al. (Reference Tsang, Heeley and Alcaino148), TRPC5 and Cav3·1 calcium channels work as a functional complex and leucine, via Cav3·1, can increase the sensitivity of POMC neurons to other satiety signals. Interestingly pharmacological activation of hypothalamic Cav3·1 reduces appetite and promotes weight loss in lean mice and enhances the efficacy of GLP1R agonism. Blouet’s group have suggested that this might benefit those patients with limited weight loss in response to GLP1R agonists(Reference Tsang, Heeley and Alcaino148).
Hunger
Less progress has been made in the identification of an aminostatic hunger response mechanism. In rodents, the mild restriction of only leucine, isoleucine, all BCAAs, methionine, or the combination of tryptophan and threonine is sufficient to reproduce the increased appetite effects observed with mild total protein/amino acid restriction supporting the concept that the dietary restriction of various amino acids triggers a ‘need’ state(Reference Khan, Spann and Münzberg149), but it was not clear if any of these effects were mediated by direct brain amino acid sensing. During rapid growth or during the postprandial stage of the diurnal feeding cycle in adults, when protein deposition in muscle and the FFM is maximally activated, it might be expected that plasma-free amino acid profiles would reflect the balance between demand and supply from amino acid absorption from food, (see Figure 8). In demand > supply circumstances, the most likely change would be either generally lower levels or a reduced ratio of indispensable to dispensable amino acids as observed after feeding a low protein diet. Karnani et al.(Reference Karnani, Apergis-Schoute and Adamantidis150) and more recently Viskaitis et al. (Reference Viskaitis, Arnold and Garau151), reported the activation of orexin/hypocretin neurons, an orexigenic neuronal population in the lateral hypothalamus, by DAA (with a relative potency order glycine > aspartate > cysteine > alanine > serine > asparagine > proline > glutamine), but with no response to leucine. Although this might suggest a potential mechanism for appetite stimulation, no influence on feeding behaviour has been established. Heeley and Blouet(Reference Heeley and Blouet152) were unable to identify any neural circuits which would explain the hyperphagia in rats fed marginally low protein diets, but did suggest that any moderation of leucine’s satiating action, (i.e. the inhibition of orexigenic NPY/AGrP neurons and the activation of anorexigenic POMC neurons), could have a net orexigenic influence.
Such lack of any direct amino acid sensing has prompted suggestions of FGF21-mediated mechanisms for the hyperphagic response to mild amino acid restriction. Since its discovery in 2000(Reference Nishimura, Nakatake and Konishi153) there has been increasing interest in this liver-derived hormone FGF21(Reference Fisher and Maratos-Flier154) and especially its action in regulating feeding behaviour, acting to increase food intake as a specific signal of dietary protein and/or amino acid restriction(Reference Hill, Berthoud and Münzberg155,Reference Hill, Qualls-Creekmore and Berthoud156) . FGF21 signalling requires both the FGF receptor 1c (Fgfr1c) and an obligatory co-receptor called β-klotho (Klb)(Reference Bookout, De Groot and Owen157). Klb is more discretely localised than Fgfr1, thereby determining the anatomical specificity of FGF21 action and early studies showed that FGF21 is involved in regulating metabolism and circadian behaviour by acting on neurones within the suprachiasmatic nucleus (SCN) of the hypothalamus and the dorsal vagal complex of the hindbrain(Reference Bookout, De Groot and Owen157). Since then as recently reviewed by Wu et al. (Reference Wu, Chaffin and Ryan158), increases in FGF21 have been reported in response to dietary restriction of branched-chain amino acids, sulfur-containing amino acids, all non-essential amino acids, and/or all essential amino acids. FGF21 is also increased following the individual dietary restrictions of methionine, leucine, threonine, and tryptophan and importantly in response to decreasing plasma glutamine after genetic manipulations in transgenic mice(Reference Cornu, Oppliger and Albert159). In terms of the neuroanatomical distribution of the FGF21 receptor complex, as assessed by β-klotho expression, whilst there is clear evidence for its occurrence in the SCN, reports of expression in other areas have not been confirmed so that according to Wu et al. (Reference Wu, Chaffin and Ryan158) the neuroanatomical distribution of Klb remains unresolved. However there are important species differences in FGF21 physiology(Reference Staiger, Keuper and Berti160) and as yet no evidence that the hyperphagic responses to amino acid restriction or low protein diets linked to elevated FGF21 observed in mice, occur acutely in humans. Furthermore, all such reported responses are dietary effects influencing hepatic FGF21 production via the portal blood supply. In order for FGF21 to be responsible for mediating changes in the circulating amino acid profile when demand > supply, hepatic FGF21 expression must be sensitive to the hepatic arterial blood supply which does account for 25 % of cardiac output but which is only one third of the portal blood supply likely to be the main influence on amino acid profiles in hepatocytes. Taken together the inclusion of a role for FGF21 in an aminostatic appetite control mechanism would seem premature.
A summary scheme for potential central aminostatic mechanisms of hunger and satiety is shown in Figure 9.
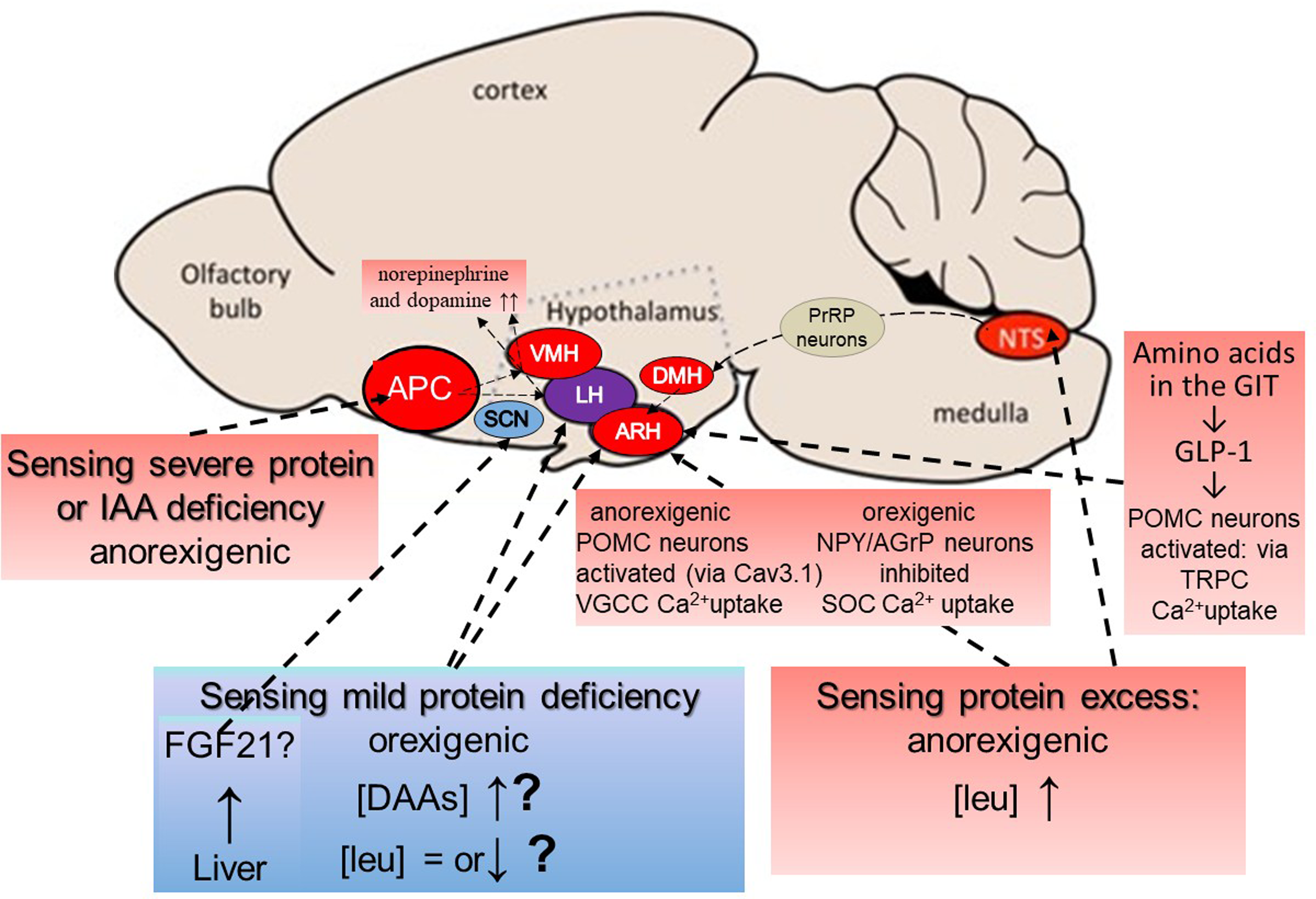
Figure 9. Potential central aminostatic mechanisms of hunger and satiety. According to Heeley & Blouet,(Reference Heeley and Blouet152) amino acid concentrations in the brain are not a simple reflection of the plasma amino acid profile but vary selectively in discrete sites under specific dietary contexts. Most is known about the anorexigenic responses to severe protein or amino acid deficiency or to excess protein. The sensing of severe protein or IAA deficiency is independent of peripheral signals and is sensed in response to rapid reductions in IAAs in the anterior piriform cortex (APC(Reference Gietzen128,Reference Gietzen129) ). Circuits connect the APC to the ventromedial and lateral hypothalamus (VMH & LH)(Reference Gietzen and Aja161) with norepinephrine and dopamine levels rapidly increased in these areas, resulting in food aversion and anorexia. The sensing of excess protein occurs in several sites. Detection of leucine in neurons in the nucleus of the solitary tract, (NTS) in the brainstem engages a circuit of NTS prolactin-releasing peptide (PrRP) neurons, connecting via the dorsomedial hypothalamus (DMH) and a population of leptin receptor-expressing neurons, to inhibit AgRP neurons in the arcuate nucleus (ARC) of the hypothalamus to suppress food intake from high-protein diets(Reference Tsang, Nuzzaci and Darwish147). Leucine sensing also occurs directly in the ARC where anorexigenic POMC neurons are activated and orexigenic NPY/AGrP neurons are inhibited through leucine regulating calcium uptake by interacting with specific calcium channel proteins(Reference Tsang, Heeley and Alcaino148). POMC activation in the ARC also occurs in response to GLP-1 released from the GIT in response to dietary amino acids, via another calcium channel protein which appears to be linked to the leucine-activated Cav3·1 calcium channel protein. The sensing of mild protein deficiency to induce hunger is the least understood aspect of aminostatic sensing. Possibilities include the activation of orexin/hypocretin neurons, an orexigenic neuronal population in the lateral hypothalamus, by dispensable amino acids(Reference Karnani, Apergis-Schoute and Adamantidis150,Reference Viskaitis, Arnold and Garau151) although this activation has not been shown to increase food intake. A lack of or moderation of postprandial leucine increase or reduction in its concentration, as observed early in the fasted state resulting in little or none of leucine‘s inhibition of orexigenic NPY/AGrP neurons and activation of anorexigenic POMC neurons, as described above, could have a net orexigenic influence(Reference Heeley and Blouet152). The hepatic secretion of FGF21 in response to dietary restriction of branched-chain amino acids, sulfur-containing amino acids, all non-essential amino acids, and/or all essential amino acids, has been shown in rodents to be associated with increased food intake(Reference Wu, Chaffin and Ryan158) with FGF21 acting on neurones within the suprachiasmatic nucleus (SCN) of the hypothalamus and the dorsal vagal complex of the hindbrain(Reference Bookout, De Groot and Owen157). However, there are important species differences in FGF21 physiology(Reference Staiger, Keuper and Berti160) and as yet no evidence that FGF21 effects observed in mice occur in humans or whether changes in the circulating amino acid profile can influence hepatic FGF21 secretion. Modified from Heeley & Bluet(Reference Heeley and Blouet152) under the CC By 4·0 licence.
Current Protein-Stat model of the regulation of the LBM by dietary protein
The current protein stat model is described in detail in Figure 10 (updated from(Reference Millward9,Reference Millward44,Reference Millward45) ).
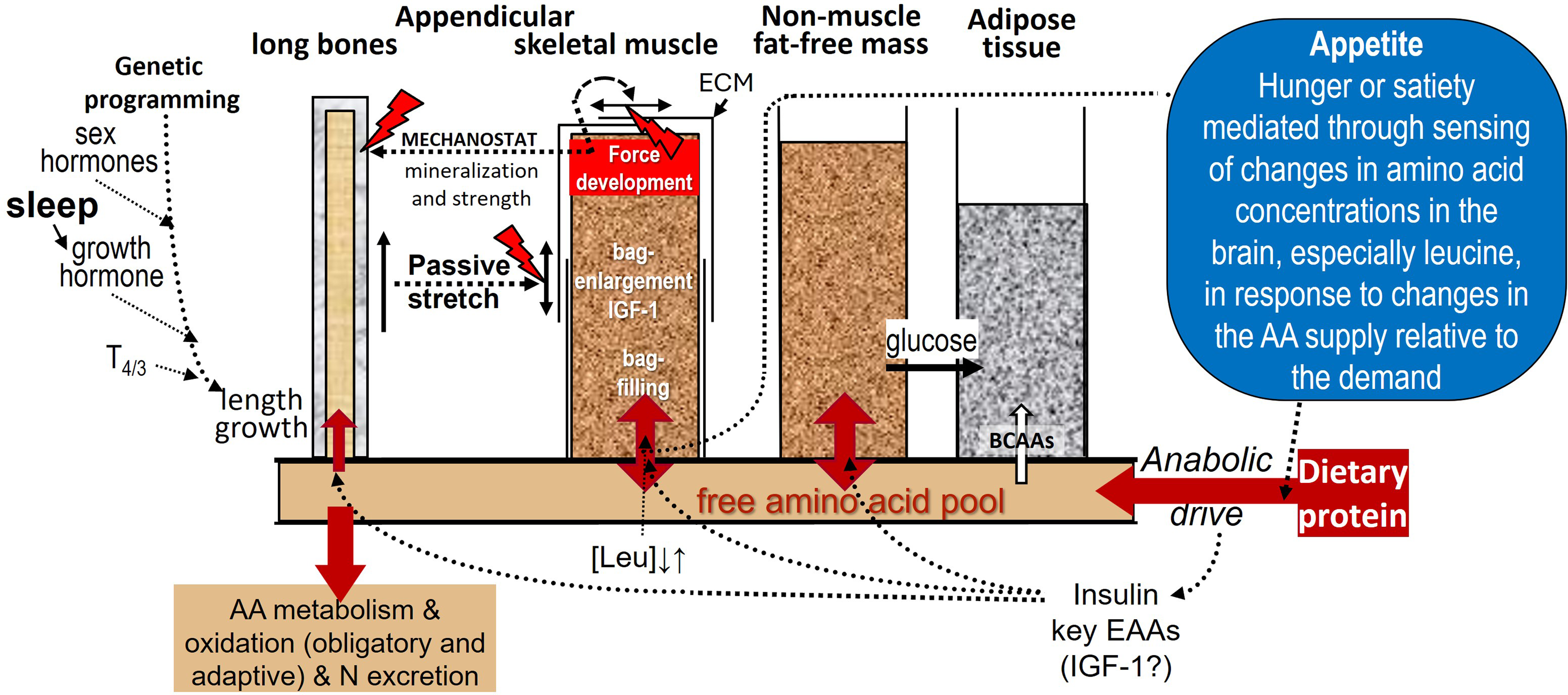
Figure 10. The Protein-Stat, (updated from(Reference Millward9,Reference Millward44,Reference Millward45) ). As described in the text, whole-body protein content is controlled through an aminostatic appetite mechanism, acting primarily to maintain skeletal-muscle mass at a level set by the linear dimensions of the organism and by the demands to manage the movement of the organism against gravity. The balance between hunger and satiety will be set by the balance between protein intake and the demand for amino acids generated mainly by the capacity of protein deposition in muscle, (as well as the demands for adaptive oxidation shown in Figure 7), with central sensors responding to circulating amino acid concentrations, especially leucine, as in Figures 8 & 9. The hierarchy of growth involves bone length driving muscle mass with growth of the non-muscle FFM not specifically regulated and driven by functional demand in response to food energy and protein intake. The interactions between dietary protein intake and its anabolic drive on bone and muscle growth are described in Figure 5. Whether the endocrine IGF-1 response to dietary protein is important for muscle growth is unclear with most known about insulin and amino acids. Muscle is shown surrounded with an extracellular matrix (ECM) which is remodelled during growth by both passive stretch allowing increases in length and by internal force development during contraction increasing cross-sectional area, the latter force acting on bone to increase mineralisation within the mechanostat concept(Reference Frost89). The linkage of bone length to muscle mass allows muscle size to be regulated at a phenotypic muscle weight–bone length ratio so that after epiphyseal closure of the appendicular long bones when adult height is achieved, muscle growth slows and eventually ceases in early adult life at the phenotypic adult size. After this muscle protein deposition only occurs within the diurnal cycle of postprandial gains which replace post absorptive losses as shown in Figures 6 and 7, or in response to force development by resistance exercise. A ‘bag full’ signal of which the molecular mechanism is poorly understood, limits protein deposition in muscle within the diurnal cycle. Any amino acid intake in excess of that required for maximal ‘bag filling’ will either expand the non-muscle FFM which is shown to be not limited in size, although regulated by functional demand, or be oxidised with the carbon skeletons leaving the liver as ketones and glucose, the latter to be taken up in adipose tissue for lipogenesis. Also, excess branched-chain amino acids can be taken up directly by adipose tissue and converted to fat. This metabolic fate of excess protein as adipogenesis is part of the Early Protein Hypothesis in which excess protein intake in infancy programmes adiposity.(Reference Koletzko, Von Kries and Monasterolo162)
There are several outstanding issues. The first is the nature of the regulatory mechanism, the ‘bag-full’ signal which limits protein deposition in muscle during the postprandial phase of the diurnal cycle of feeding and fasting (as in Figure 6).
The demonstration of the ‘bag-full’ phenomenon with studies of postprandial MPS in adults(Reference Bohé, Aili Low and Wolfe95–Reference Mitchell, Phillips and Hill98) confirmed a regulatory signal which limited protein deposition in the myofibre. However, its mechanism in terms of the signalling examined in those studies was by no means resolved. In fact, MPS fell whilst intracellular leucine concentration remained elevated and mTORC1 signalling was still active(Reference Atherton, Etheridge and Watt96,Reference Mitchell, Phillips and Hill98) at least those signals associated with the initiation of protein synthesis (p70S6K1, and 4EBP1). However eEF2 phosphorylation does seem to increase coincident with the fall in MPS(Reference Mitchell, Phillips and Hill98) and this indicates that the elongation phase of protein synthesis became inhibited, an effect observed in meal-fed rats in which the increased MPS in response to a meal was transient returning to baseline coincident with an increase in eEF2 phosphorylation(Reference Norton, Layman and Bunpo163). Classically, eEF2 phosphorylation increases in response to AMPK signalling associated with energy stress, such as muscle contraction, to protect ATP and PCr levels since it is the translational phase of MPS which is so energy dependent(Reference Proud164). In meal-fed rats, post meal supplementation with either leucine or carbohydrate prevented eEF2 phosphorylation and extended the increase in MPS(Reference Wilson, Layman and Moulton165). However, in human muscle, the ‘bag-full’ return to baseline of MPS occurs with no indication of inhibitory AMPK signalling or energy stress in terms of muscle ATP or phosphorylcreatine concentrations(Reference Mitchell, Phillips and Hill98). This means that the triggering of the response of eEF2 was unexplained. Nevertheless, it represents the only observed response which could be involved in mediating the ‘muscle-full’ inhibition of MPS.
It was originally assumed that anabolic/catabolic signals associated with changes in muscle cell volume would be important for the ‘bag-filling-bag-full’ signalling concept(Reference Häussinger, Gerok and Roth166). As extensively discussed elsewhere(Reference Millward45), it is highly likely that myofibre volume changes occur during the feeding-fasting cycle, with evidence for linkage between cell volume, mTOR and two different anion channels associated with cell swelling(Reference Kumar, Xie and Ta167) and shrinkage(Reference Gosmanov, Lindinger and Thomason168). However, after reviewing the available evidence a satisfactory mechanism linking volume change to the inhibition of MPS was not immediately obvious.
Taken together, the above indicates that while the ‘bag-full’ termination of postprandial protein deposition in muscle has clearly been shown to occur, its mechanism remains to be identified. As for bag enlargement, this is an inevitable accompaniment to muscle growth. As discussed elsewhere(Reference Millward9) increased muscle collagen synthesis is an early response to stretch-overload-induced muscle growth in the fowl(Reference Laurent, Sparrow and Bates65), and is observed in adult muscle stimulated by various intense exercise regimes which induce muscle enlargement(Reference Millward and Smith47,Reference Brightwell, Latham and Thomas169) . ECM remodelling is part of the satellite cell activation and myogenesis likely to accompany the muscle hypertrophy in response to maximal muscle contractions (see(Reference Millward9)). During the rapid developmental muscle growth in young rodents, it would be predicted that there is continuous remodelling of the ECM as indicated by the active proteoglycan synthesis in muscle during rapid growth which was reduced as growth slowed with parallel changes in muscle IGF-1 shown in Figure 4. This is an area where more work is warranted.
Another unresolved issue is the nature of human bone growth, i.e. the extent that it is saltatory, (discontinuous with leaps), involving periods of growth and stasis in human height(Reference Lampl and Johnson170,Reference Lampl and Schoen171) and animal length(Reference Noonan, Farnum and Leiferman172). Although this is not accepted by all, nevertheless when considered in the context of the mechanotransduction of muscle growth, saltatory growth implies that passive stretch forces associated with a saltation could be much greater than those implied by a continuous but very slow rate of human long bone growth. Indeed it might be argued that bursts of rapid growth in length are necessary to achieve sufficient passive stretch to transduce increased myofibre length growth through sarcomere additions. This raises the possibility of fibre damage occurring in response to saltations, a phenomenon which could explain the anecdotal ‘growing pains’ reported in young children(Reference Lehman and Carl173), and lambs(Reference Noonan, Farnum and Leiferman172).
Is the Protein-Stat model relevant to better understanding childhood growth? The key implication of the growth model for childhood, is the concept of a growth capacity for skeletal muscle, especially for the appendicular muscles, which is set by mechanotransductive forces. These forces can derive from both an increase in stature or increased demands for muscle work, but when they are minimal, which may be quite often in preschool children, dietary stimulation of muscle growth is likely to be very limited apart from the diurnal repletion of postabsorptive losses as shown in Figures 6 and 7. The inability of dietary protein alone to stimulate muscle growth in childhood in excess of the growth which accompanies either height growth or in response to excessive physical demands, is one explanation of the ‘Early Protein Hypothesis’(Reference Koletzko, Von Kries and Monasterolo162). This postulates that dietary protein in excess of the intake required for lean tissue growth, a demand which may be quite low after the first year of life, increases plasma insulin and IGF-1 concentrations. This response mediates adipogenic activity and associated weight gain, and the programming of obesity in later childhood., and implies that the endocrine action of IGF-1 has no influence on muscle growth during stasis periods for bone length growth. Similarly, during adult maintenance, the phenotypic muscle mass can be maintained over a wide range of dietary protein intakes, with no evidence that the phenotypic lean body mass is a function of dietary protein intake, apart from that which varies with BMI and weight gain(Reference Millward, Truby and Fox174). In this latter case, the FFM accounts for 40 % and 27 % of the excess weight associated with increasing BMI in men and women respectively. Whilst the anatomical site of this excess FFM in the obese has not been unequivocally identified, its sexual dimorphism, like that of skeletal muscle and the overall FFM in non obese adults, suggests that some of it includes increased muscle mass(Reference Millward, Truby and Fox174).
Finally, the role of amino acid sensing mechanisms linking the regulation of appetite and protein intake to the metabolic demand for amino acids is becoming clearer especially the role of leucine, with its importance in regulating mTORC1 and proteostasis(Reference Wolfson, Chantranupong and Saxton175–Reference Blouet, Ono and Schwartz179), extended to regulating key appetite-related neurons, at the level of the regulation of calcium channel proteins mediating neuronal calcium uptake(Reference Tsang, Heeley and Alcaino148). Whilst it is clear that this can explain satiety it remains to be demonstrated that such action can also influence hunger but the intensity of current research efforts in this area, as demonstrated by the results shown in Figure 9, should reveal answers to this question.
Conclusions
The work described here represented my main scientific interest and involved a programme of experimental work which was largely complete by the early 2000s to which I returned in the last few years attempting to reconcile and update our findings to align them with the current state of the art. To be able to do this is a privilege which comes with retirement. My time at Surrey did include other important work with students and colleagues and external agencies unrelated to protein and the Protein Stat but which would fall within the remit of the Blaxter award and which was similarly rewarding. This has not been referred to here because it involves different stories. I am extremely grateful to those colleagues involved in those programmes and hope an opportunity to tell those stories will occur in the not-too-distant future.
Dedication
In addition to acknowledging the intellectual and practical support of those identified below, I would like to dedicate this paper to the memories of John Waterlow, Peter Garlick, Peter Sender, Peter Reeds, John Rivers, Philip Payne, Mike Rennie, Dave Halliday and to Paul Pacy when he was well, each of them sources of inspiration and pioneers in the development of the field.
Acknowledgements
Of the many colleagues on whose wisdom I have drawn I would particularly like to thank Ann Ashworth-Hill, Mike Golden and Alan Jackson for sharing their lifetimes experience of severe childhood malnutrition. At the LSHTM the remarkable output of the CNMU (of which only that of my own group is referred to here), could not have been achieved without the support of Jutinder Ryatt, Dickson NNaneyelugo, Philip Broadbent, Malcolm Cox, and above all Peter Bates’ expertise, enabling much of my own experimental work and that of my students, George Grimble, Geof Laurent, John Brown, Rudolfo Giugliano, Margaret Jepson, Penny Coyer, Asma Omer, Sana Janakat, Zainal Yahya, and Post Docs, Bhanu Odedra and Jenny Pell. Our human work, initially with Richard Edwards, Dave Halliday, Mike Rennie and Steve Wolman, was enabled by an international collaboration with Denny Bier and Dwight Matthews, providing groundbreaking stable isotope expertise, with our subsequent work on nitrogen homeostasis and PPU, led by Paul Pacy with students Gill Price, Marcello Quevedo, Neil Gibson and Amelia Fereday. This was enabled by the skills of Malcolm Cox at the CNMU and by Eric Ah-Sing at Surrey. There, I was able to draw upon the expertise in the regulation of appetite and the entero-pancreatic axis of Linda Morgan and students, Sam Long and Wendy Hall. Christine Williams, and Bruce Griffin introduced me to lipids and with John Wright, Karen Slevin, Nahed Hussein, Margaret Griffin and Ian Davies, we were able to explore insulin resistance, fatty acid metabolism, and in collaboration with Tom Sanders at KCL, dietary n-3 PUFAs and atherogenic risk. With Helen Truby, Linda Morgan, Bruce Griffin and Kath Hart, we were able to conduct the BBC-funded diet trials obesity management RCT. Finally, Sue Lanham-New together with Andrea Darling afforded me the opportunity to contribute to their research into dietary protein and bone health. I particularly want to thank Sue for encouraging me to apply for this award.
Financial support
No financial support was received for the preparation of this paper.
Competing interests
There are no conflicts of interest.












 Open access
Open access