


Introduction
Catatonia is a neuropsychiatric disorder that occurs in the context of a range of psychiatric and general medical disorders (Fink & Taylor, Reference Fink and Taylor2003; Oldham, Reference Oldham2018). Common clinical features include mutism, stupor, social withdrawal, posturing, and stereotypes (Dawkins et al., Reference Dawkins, Cruden-Smith, Carter, Amad, Zandi, Lewis, David and Rogers2022). Catatonia has a population incidence of approximately 10 per 100,000 person-years (Rogers et al., Reference Rogers, Watson, Badenoch, Cross, Butler, Song, Hafeez, Morrin, Rengasamy, Thomas, Ralovska, Smakowski, Sundaram, Hunt, Lim, Aniwattanapong, Singh, Hussain, Chakraborty and Rooney2021) and is associated with increased mortality (Cornic et al., Reference Cornic, Consoli, Tanguy, Bonnot, Périsse, Tordjman, Laurent and Cohen2009; Niswander, Reference Niswander1963; Puentes, Brenzel, & De Leon, Reference Puentes, Brenzel and De Leon2017), and a range of medical complications (Funayama et al., Reference Funayama, Takata, Koreki, Ogino and Mimura2018). Alongside treating the underlying condition, the mainstay of treatment for catatonia – regardless of etiology – consists of benzodiazepines or electroconvulsive therapy (ECT) (Denysenko et al., Reference Denysenko, Sica, Penders, Philbrick, Walker, Shaffer, Zimbrean, Freudenreich, Rex, Carroll and Francis2018; Rogers et al., Reference Rogers, Pollak, Begum, Griffin, Carter, Pritchard, Broadbent, Kolliakou, Ke, Stewart, Patel, Bomford, Amad, Zandi, Lewis, Nicholson and David2023). It remains unclear to what extent underlying cause correlates with catatonic signs (Dawkins et al., Reference Dawkins, Cruden-Smith, Carter, Amad, Zandi, Lewis, David and Rogers2022).
The observation that mental illness is commonly familial was made in the Hippocratic Corpus as far back as the 5th century BC (Evans, McGrath, & Milns, Reference Evans, McGrath and Milns2003). Twin studies of disorders including alcoholism, depression, and schizophrenia in the 20th century subsequently confirmed these disorders had a heritable component, albeit to varying degrees (Kendler, Reference Kendler2001). More recently, genome wide association studies (GWAS) have endeavored to identify specific genetic variants linked with psychiatric disorders. Even with large sample sizes, single nucleotide polymorphisms (SNPs) identified in existing GWAS account for only a small proportion of the heritability estimated from twin studies, which may be accounted for by other variant types, inflated results from twin studies, or the interaction with other genes and environmental factors (Andreassen, Hindley, Frei, & Smeland, Reference Andreassen, Hindley, Frei and Smeland2023). Notwithstanding these limitations, modern psychiatric genetic research has advanced clinical translation by identifying risk genes, polygenic risk scores, and shared biological pathways; enabling early risk prediction and personalized care (Smoller et al., Reference Smoller, Andreassen, Edenberg, Faraone, Glatt and Kendler2019; Wray et al., Reference Wray, Ripke, Mattheisen, Trzaskowski, Byrne, Abdellaoui, Adams, Agerbo, Air, Andlauer, Bacanu, Bækvad-Hansen, Beekman, Bigdeli, Binder, Blackwood, Bryois, Buttenschøn, Bybjerg-Grauholm and Sullivan2018). Pharmacogenomics such as CYP450 and HLA gene insights optimize medication selection and dosing (Bousman & Hopwood, Reference Bousman and Hopwood2016).
The major psychiatric diagnostic manuals (ICD-11 and DSM-5-TR) now recognize that catatonia is not necessarily part of schizophrenia. Although this has rendered the diagnostic process in catatonia more valid, it leaves the clinician with the difficult task of ascertaining which of many potential etiologies is most relevant. In some case series, there remain cases of catatonia that –despite extensive investigation – do not have a clear cause (Espi Forcen et al., Reference Espi Forcen, Respino, Eloge, Baldwin, Burns, Patron and Hebert2022; Zingela et al., Reference Zingela, Stroud, Cronje, Fink and Van Wyk2022). It is in this context that there is increasing interest in genetic disorders underlying catatonia (Raffin et al., Reference Raffin, Consoli, Giannitelli, Philippe, Keren, Bodeau, Levinson, Cohen and Laurent-Levinson2018). Making a genetic diagnosis in catatonia could be important for ascertaining prognosis, guiding investigations for other features of a particular syndrome, and for certain conditions accessing disease-modifying therapies. Currently, however, no comprehensive synthesis of genetic abnormalities in catatonia exists in the published literature to our knowledge.
We therefore aimed to conduct a systematic review to identify the genetic abnormalities reported in catatonia, their frequency, and – where possible – the strength of any association. For completeness, neuropsychiatric comorbidities are also reported. Due to the anticipated heterogeneity of the literature, we planned to conduct a narrative synthesis without meta-analysis.
Methods
We carried out a systematic review, pre-registered on PROSPERO (CRD42023455118). The PRISMA 2020 checklist is shown in Supplementary Table 1.
Eligibility criteria
Due to the anticipated paucity of large rigorous epidemiological studies, eligibility criteria were kept broad, as shown in Table 1.
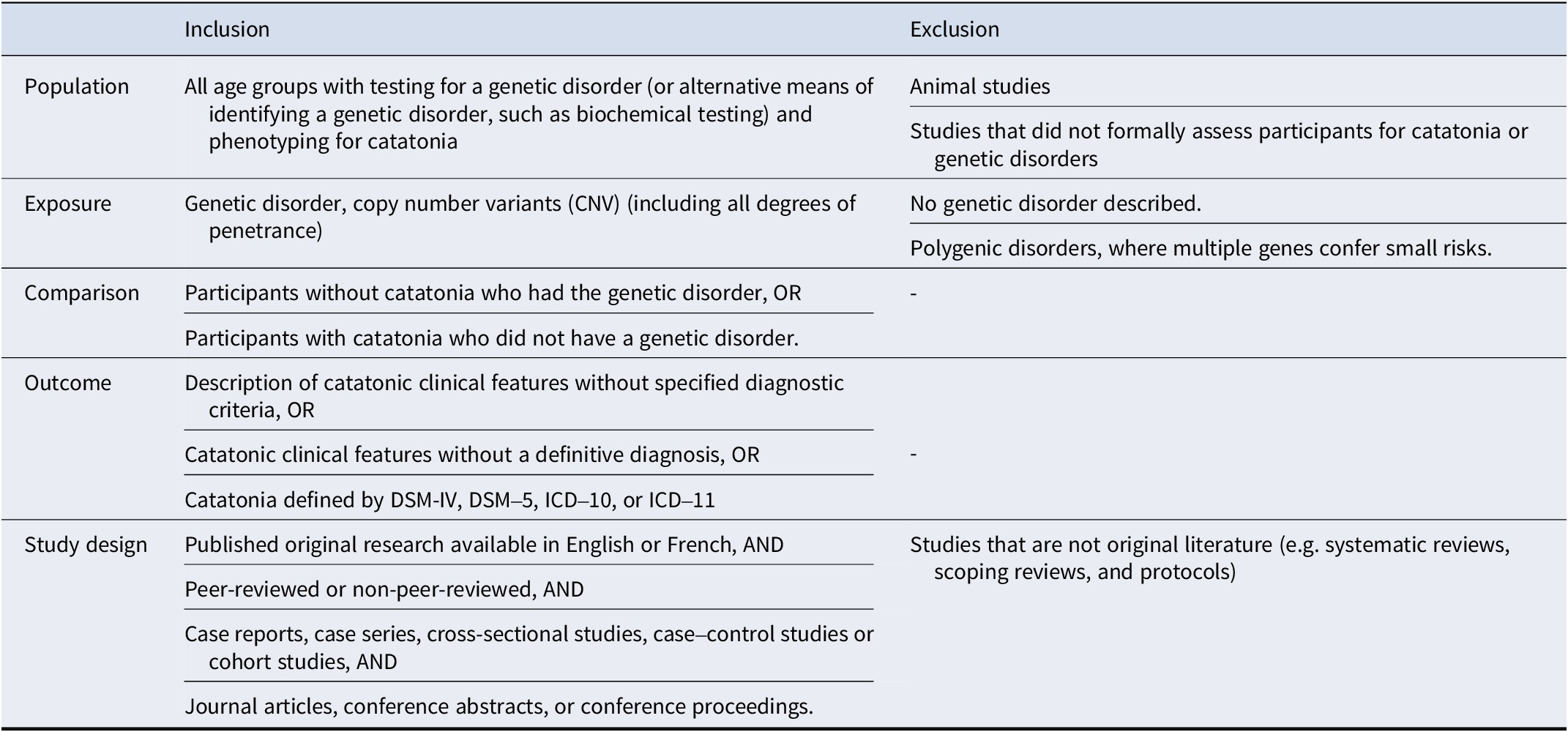
Table 1. Eligibility criteria
Search strategy
Searches were conducted on Ovid using MEDLINE All, Embase classic + Embase, PsycINFO, and AMED from inception to August 15, 2023. References were also screened for relevant studies. The search strategy was limited to human studies using search keywords and structured vocabulary including but not limited to ‘catatonia’, ‘gene’, ‘polymorphism’, ‘genome’. The full search terms can be found in Supplementary Table 2. Pre-prints without peer review were not included to ensure the quality of included data. Duplicates were removed automatically on Ovid and subsequently identified manually. Where studies used the same population, but reported different genetic tests, results for each genetic test were included.
Eligibility was initially screened by title and abstract by two authors (KR & IC) independently with blinding using the Covidence software. Where there was disagreement between authors, the article was retained for further assessment. The same procedure was used to evaluate full texts, but a third author (JPR) arbitrated where there were disagreements.
Data extraction
Data were extracted independently with blinding from eligible full-text articles by a group of authors (IC, KR, JBF, JB, ER, BC, and CW). Where discrepancies occurred between the two original extractors, a third author arbitrated. If additional information was required, the corresponding author of the relevant manuscript was contacted. The PRISMA flowchart can be found in Supplementary Table 3.
Data were collected on the following study characteristics: paper metadata (title, author, citation), demographic characteristics of the sample, country of study population, data collection period, study population, single- vs. multicenter, study design, genetic testing, genetic abnormality, catatonia definition, catatonic signs, and treatment response.
Outcomes were defined as any catatonic features (with or without formal diagnostic criteria) in populations with genetic disorders. Catatonic features were extracted where data were available. Where populations had a defined genetic syndrome with an established single genetic mutation, the genetic syndrome was reported even if individuals had not had genetic analysis.
Quality assessment
The Joanna Briggs Institute tools were used to assess cross-sectional, case report, case series, case control, and cohort designs. Cohort studies without a comparison group were assessed using the case series tool. Authors (IC, KR, JB, JPR, JBF, CW, and ER) independently assessed papers with a third author acting as arbitrator (BC or JB). Risk of bias was categorized as low, moderate, or high risk using the following cut-offs: Case reports rated out of 7 points were low (6–7), moderate (3–5), high (0–2). Case series and case–control studies rated out of 10 points were low (8–10), moderate (4–7), high (0–3) risk of bias. Cohort studies rated out of 11 points were low (8–11), moderate (4–7), high (0–3) risk of bias.
Data analysis
Demographic information of participants with each genetic condition was reported in addition to the median number and duration of catatonic episodes. Due to the clinical and methodological heterogeneity between studies, a narrative approach was used to synthesize the data. Association studies (where a comparison group was presented) were considered separately from studies without comparison groups. Additional information was provided for genetic conditions where 10 or more cases of catatonia were reported in the literature. The number of individuals with specific catatonic signs, as well as the response to treatment for individuals with catatonia was reported separately.
Role of the funding source
The funding sources had no part in the study design, data collection, analysis, or writing of the manuscript or the decision to submit it for publication.
Results
Description of included studies and individuals
The systematic literature searches identified 2488 articles, of which 99 were included in the systematic synthesis, as shown in Supplementary Figure 3. A full list of included studies with their characteristics is shown in Supplementary Table 4. Study quality was good in 29 studies, moderate in 53, and poor in 17, as shown in Supplementary Tables 5a–d.
Articles were published between 1972 and 2023. Of the 99 included studies, there were 9 cohort studies, 18 case–control studies, 18 case series, and 54 case reports. There were 13 multicenter studies and 86 single-center studies. There were 84 peer-reviewed studies and 15 studies published without peer review, often as conference abstracts or proceedings. In terms of country, 28 studies were conducted in the USA, 17 in Germany, 12 in France, 5 in Italy, 5 in Japan, and 32 from 17 other countries. In terms of funding, 15 studies reported no funding, 22 studies reported only non-commercial funding, 4 studies reported commercial funding, and 58 studies had no statement on funding.
Catatonia outcomes were reported for 8600 individuals, of whom 636 (7.4%) had catatonia and a genetic abnormality, and 157 (1.8%) had catatonia without a genetic abnormality. A total of 2385 (27.7%) had a genetic abnormality without catatonia and 476 (5.5%) had neither catatonia nor a genetic abnormality. Sex was reported for 6080 individuals, of whom 3208 (52.8%) were male and 2872 (47.2%) female. Age at onset of first catatonic episode ranged between 4 and 80 where it was given for (n = 43) individuals and ranged from a mean of 28.8 (SD 16.3) to 38.6 (SD 12.5) where it was provided for groups. Ethnicity was reported in 1563 individuals, of whom 531 were White, 1028 Asian, 3 Black, and 1 Mixed.
Diagnosis of catatonia was made using DSM-IV criteria (17) in 3072 individuals, DSM-5 criteria (18) in 1685, ICD-10 criteria (19) in 608, and other specified criteria in 396, while a diagnosis was made without any specified criteria in 2293, and 145 individuals presented with catatonic features without an explicit diagnosis of catatonia. The number of catatonic episodes was available in 57 individuals, ranging from 1 to 4 (median 1, IQR 1 to 2). Of these, 28% (16/57) had 2 or more catatonic episodes. Duration of the index catatonic episode was available in 35 individuals, ranging from 4 to 2920 days (median 180.0, IQR 37.5 to 667.5). The genetic diagnosis was made by comparative genomic hybridization (CGH) in 11 individuals, exome sequencing in 21, fluorescence in situ hybridization (FISH) in 7, karyotype in 10, microarray in 985, PCR in 3445, whole genome sequencing in 4, other genetic techniques in 266, and through clinical diagnosis alone in 1; diagnostic technique was not stated in 3454 individuals.
Association studies
Of the 18 studies examining the association between catatonia and a specific genetic abnormality, 10 were group-level association studies, 3 were group-level linkage, 4 were segregation analyses, and 1 was factor analysis. There were 15 case–control studies, 1 cohort study, and 2 case series. Among these studies, 3 were at low risk of bias, 12 at moderate risk of bias, and 3 at high risk of bias. A particular issue with risk of bias was a lack of appropriate strategies for dealing with confounding.
Fourteen (78%) of the association studies were conducted in individuals with periodic catatonia, a rare subtype of catatonia characterized by recurrent episodic catatonia (Stöber et al., Reference Stöber, Saar, Rüschendorf, Meyer, Nürnberg, Jatzke, Franzek, Reis, Lesch, Wienker and Beckmann2000). Stöber, Saar, et al., (Reference Stöber, Saar, Rüschendorf, Meyer, Nürnberg, Jatzke, Franzek, Reis, Lesch, Wienker and Beckmann2000) provided the first genome wide linkage study of periodic catatonia in 12 multiplex pedigrees, identifying linkage with areas of chromosome 15q15 and suggesting other areas of linkage on the long arm of chromosome 22. Stöber et al. (Reference Stöber, Seelow, Rüschendorf, Ekici, Beckmann and Reis2002) replicated this linkage on chromosome 15q15 in a new set of four multiplex families; placing the susceptibility region to an interval between marker D15S1042 and D15S659. Stöber et al. (Reference Stöber, Kohlmann, Siekiera, Rubie, Gawlik and Möller-Ehrlich2005) further examined candidate genes on chromosome 22q. Systematic mutation screening in individuals from chromosome 22q-linked pedigrees identified 7 SNPs at KIAA0767/DIP locus which did not co-segregate with the disease. Seventeen SNPs at KIAA1646/CERK were identified, none of which co-segregated.
Candidate genes at chromosome 22q13 have also been investigated owing to the high prevalence of catatonia in Phelan-McDermid syndrome (PMS), characterized by abnormalities in the SHANK3 gene in this region (Kohlenberg et al., Reference Kohlenberg, Trelles, McLarney, Betancur, Thurm and Kolevzon2020). In a sample of 620 individuals (212 chronic schizophrenia; 56 periodic catatonia; 106 bipolar disorder; 284 controls) Selch et al. (Reference Selch, Strobel, Haderlein, Meyer, Jacob, Schmitt, Lesch and Reif2007) found associations between two intronic SNPs (rs2235349 and rs2076137) of the MLC1 gene located on chromosome 22q13.33, and periodic catatonia. Candidate genes on chromosome 22q13.3 have previously been implicated in periodic catatonia by Meyer et al. (Reference Meyer, Huberth, Ortega, Syagailo, Jatzke, Mössner, Strom, Ulzheimer-Teuber, Stöber, Schmitt and Lesch2001), demonstrating co-segregation with a WKL1 Leu309Met missense variant in an extended pedigree. However, a later study by McQuillin et al. (Reference McQuillin, Kalsi, Moorey, Lamb, Mayet, Quested, Baker, Curtis and Gurling2002) was unable to replicate this association in a population of 174 schizophrenia cases. They identified an insertion/deletion, one-missense substitution, and two synonymous substitutions in exon 11. None of these polymorphisms segregated with schizophrenia despite being thought to alter WKL1 protein structure. Another candidate gene located on chromosome 22q13.33 examined by Gross et al. (Reference Gross, Grimm, Ortega, Teuber, Lesch and Meyer2001) was the cadherin gene CELSR1. Seventeen allelic variants were identified, none of which co-segregated with catatonic schizophrenia in a pedigree of 5 family members.
Following Stober’s early work, several studies have investigated putative candidate genes at chromosome 15q15 in periodic catatonia. The DLL4 gene at15q15, which produces a NOTCH 4 ligand, was investigated by McKeane et al. (Reference McKeane, Meyer, Dobrin, Melmed, Ekawardhani, Tracy, Lesch and Stephan2005). In a case–control study of 3 affected individuals, no SNPs were found which co-segregated with disease in the extended pedigree. Screening for genetic variants of SLC30A4 at chromosome 15q15–21 has also been attempted (Küry et al., Reference Küry, Rubie, Moisan and Stöber2003). Despite demonstrating that a critical region in this zinc transporter gene (D15S1042 to D15S659) perfectly co-segregated with individuals with periodic catatonia, no pathogenic variants in either coding or promoter regions were found to explain the phenotype. Most recently Schanze et al. (Reference Schanze, Ekici, Pfuhlmann, Reis and Stöber2012) evaluated conserved non-genic sequences on chromosome 15q15 at the PLCB2 and DLL4 loci. Three variants in conserved and ultra-conserved sequences were found only in cases of periodic catatonia. However, significance could not be confirmed in a case–control association study. Meyer (Reference Meyer2002) examined the CX36 gene on chromosome 15q14; encoding a gap-junction protein expressed in brain and retina tissue. Exon sequencing identified 3 polymorphic sites, none of which co-segregated with catatonic schizophrenia.
Three studies examined trinucleotide repeat expansions at putative candidate loci for periodic catatonia (Bengel et al., Reference Bengel, Balling, Stöber, Heils, Li, Ross, Jungkunz, Franzek, Beckmann, Riederer and Lesch1998; Lesch et al., Reference Lesch, Stöber, Balling, Franzek, Li, Ross, Newman, Beckmann and Riederer1994; Stöber, Saar, et al., Reference Stöber, Saar, Rüschendorf, Meyer, Nürnberg, Jatzke, Franzek, Reis, Lesch, Wienker and Beckmann2000). Polymorphic B33 CTG repeat locus on chromosome 3 was assessed in 45 patients with periodic catatonia and 43 control subjects, with no significance identified (Bengel et al., Reference Bengel, Balling, Stöber, Heils, Li, Ross, Jungkunz, Franzek, Beckmann, Riederer and Lesch1998). The B37 CAG repeat locus on chromosome 12 was investigated in patients with periodic catatonia and controls; again without significance (Stöber, Saar, et al., Reference Stöber, Saar, Rüschendorf, Meyer, Nürnberg, Jatzke, Franzek, Reis, Lesch, Wienker and Beckmann2000). Also examined were CAG repeats on exome 1 of the hKCNN3 gene, coding the human calcium-activated potassium channel on chromosome 1q21. Using 12 periodic catatonia pedigrees, no linkage was identified (Lesch et al., Reference Lesch, Stöber, Balling, Franzek, Li, Ross, Newman, Beckmann and Riederer1994).
Genetic conditions with 10 or more cases of catatonia reported
Three genetic conditions had at least 10 cases of catatonia reported in the literature: PMS (80 cases), 22q11.2 deletion syndrome (DiGeorge syndrome) (23 cases), and Down’s syndrome (19 cases). Of the 26 relevant studies, there were 4 cohort studies, 10 case series, 12 case reports, and no case–control studies. There were 10 studies at low risk of bias, 13 at moderate risk, and 3 at high risk. Particular issues were lack of clear inclusion criteria for cases, and limited information about interventions given. A summary of the results for each condition is shown in Table 2.
Table 2. Genetic conditions with 10 or more cases of catatonia reported
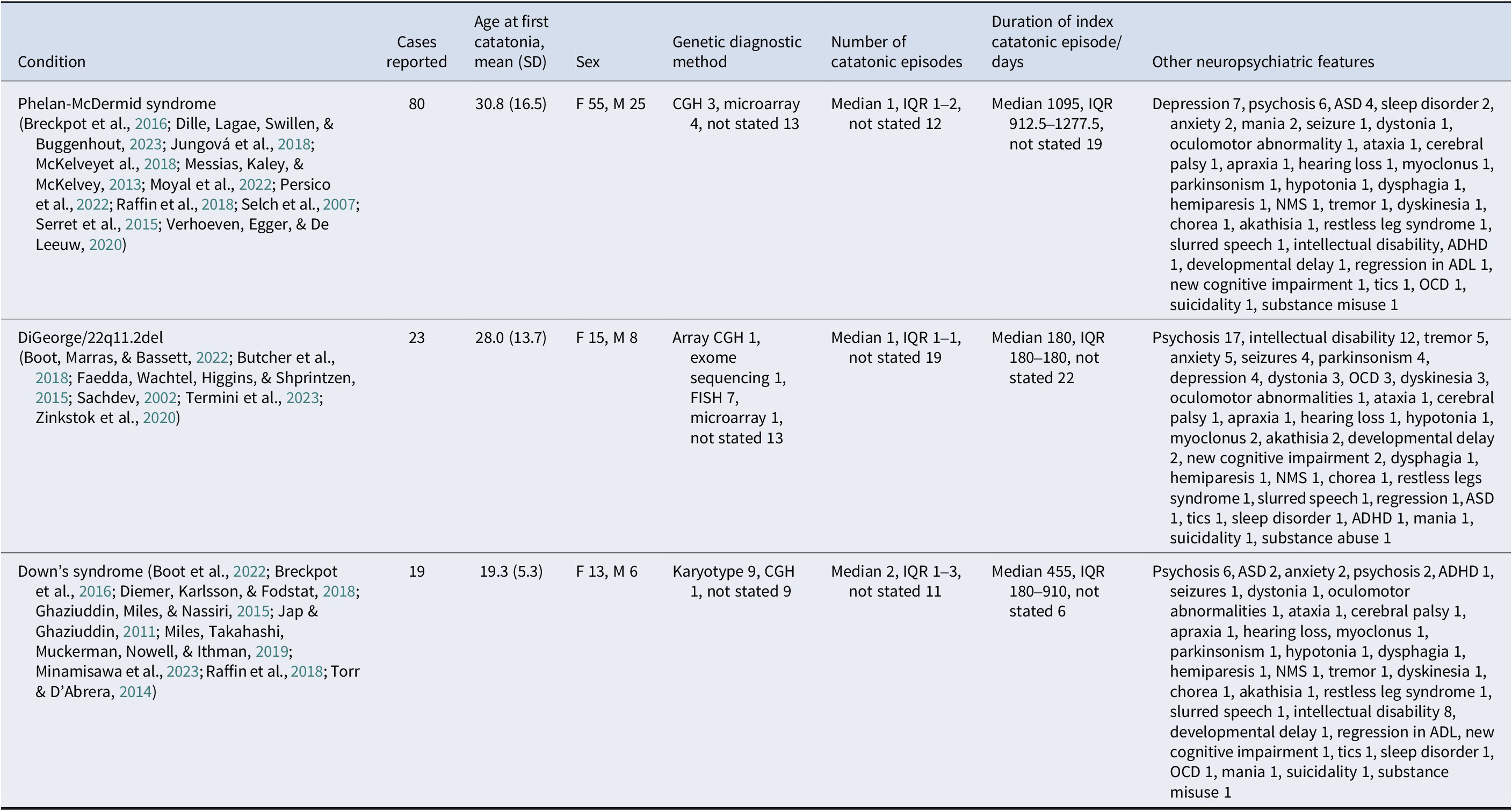
Abbreviations: ADHD – attention deficit hyperactivity disorder. ADL – activities of daily living. ASD – autism spectrum disorder. NMS – neuroleptic malignant syndrome. OCD – obsessive-compulsive disorder.
Genetic conditions with fewer than 10 cases of catatonia reported
Forty-four genetic conditions had fewer than 10 cases reported with the results shown in Table 3. Of the 40 relevant studies, there were 4 cohort studies, no case–control studies, 6 case series, and 30 case reports. There were 15 studies at low risk of bias, 19 at moderate risk, and 6 at high risk. Particular issues were inadequate reporting of case demographics and histories, and lack of reporting on adverse events resulting from interventions. A summary of the results for each condition is shown in Table 3.
Table 3. Genetic conditions with fewer than 10 cases of catatonia reported
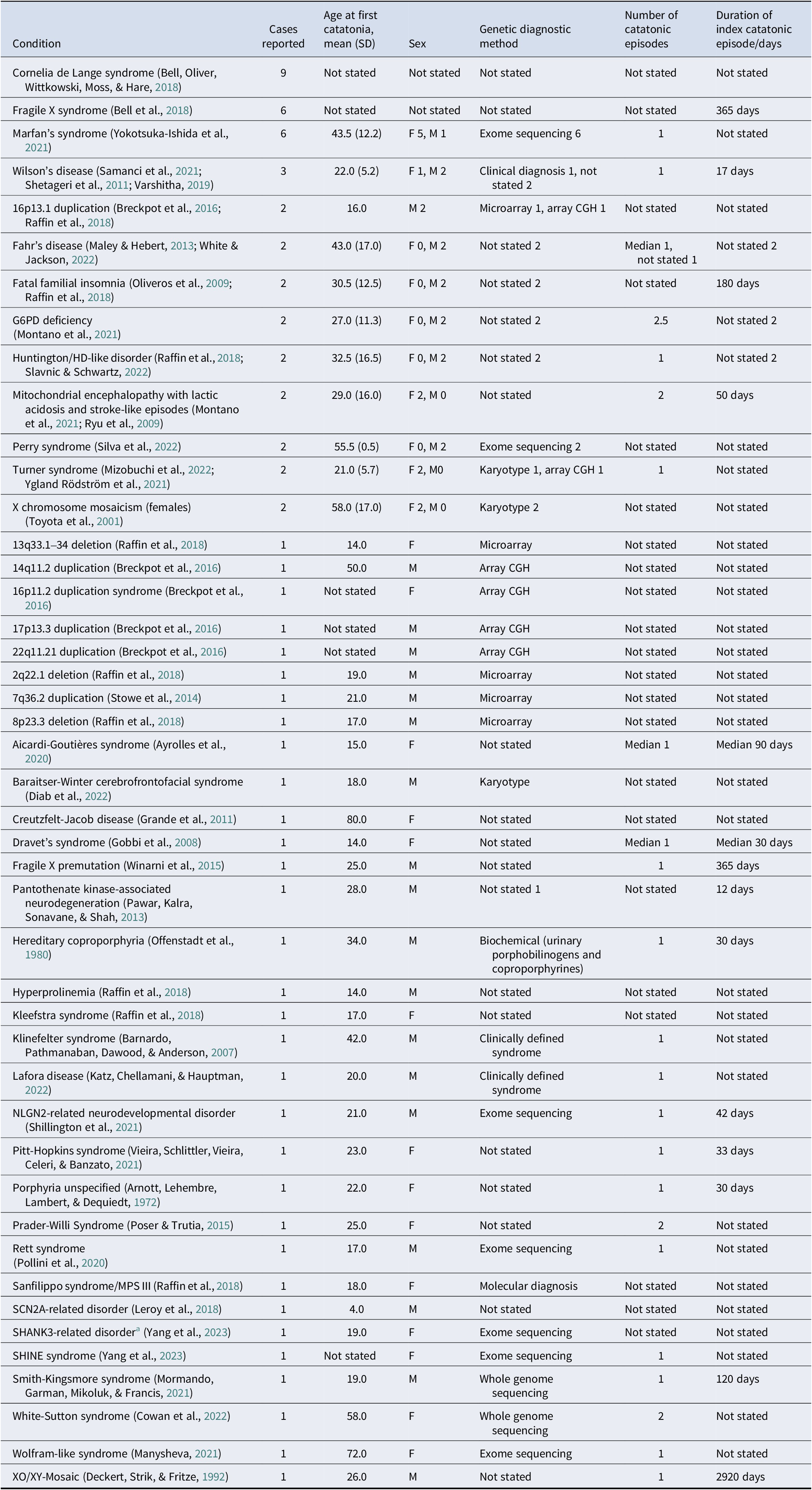
Abbreviations: MPS-III – mucopolysaccharidosis type 3. SHINE – short stature, hyperextensibility, intellectual disability, nail abnormalities, ear abnormalities.
a Not clearly Phelan-McDermid syndrome
Specific catatonic signs
Among 574 individuals with catatonia who had a genetic condition, 211 had some information on the specific clinical signs that they displayed, as illustrated in Figure 1. Of the 52 relevant studies, there were 2 cohort studies, 11 case series, and 39 case reports. 19 studies were at low risk of bias, 27 at moderate risk, and 6 at high risk.
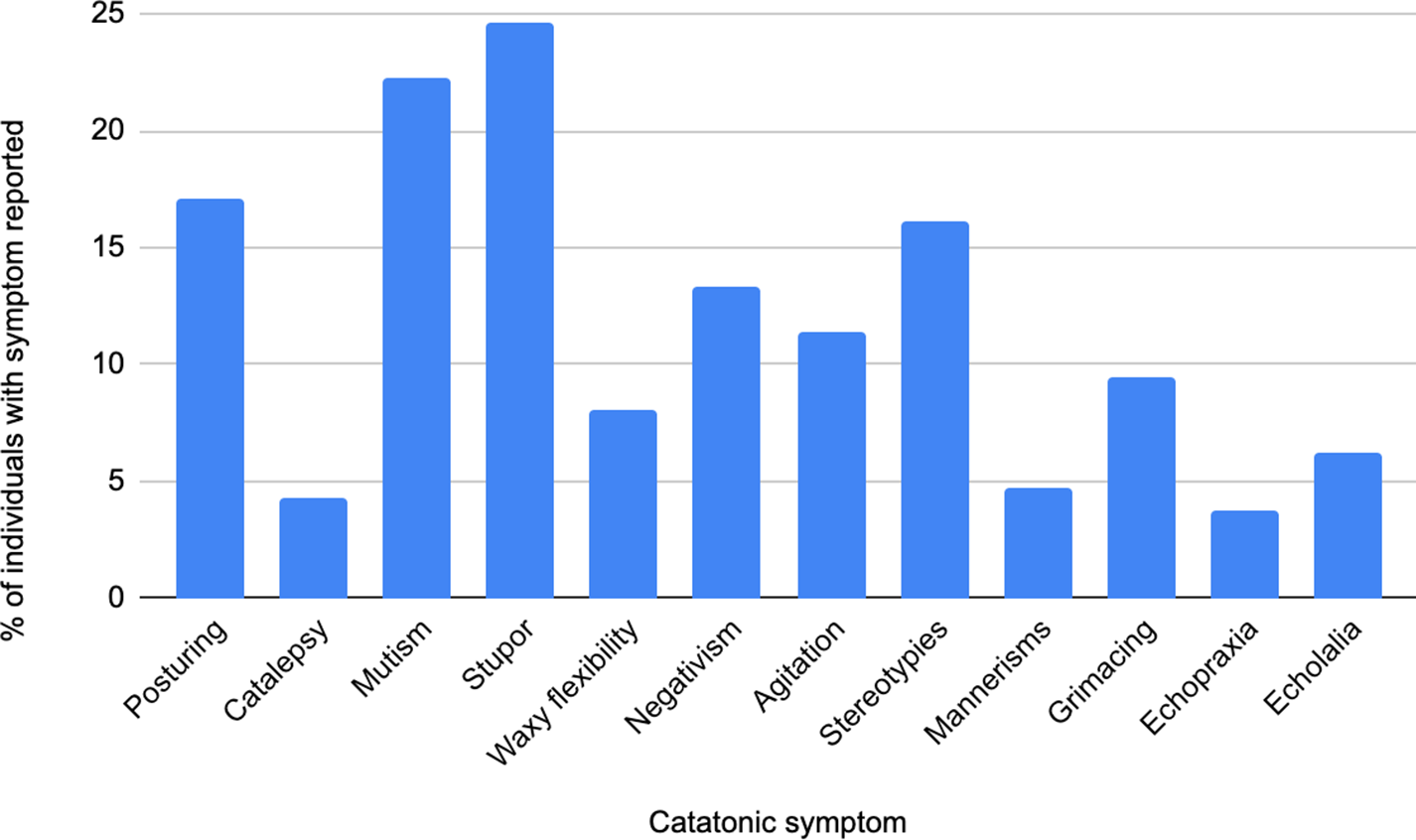
Figure 1. Bar chart showing catatonic signs present in individuals with catatonia and a genetic condition.
Treatment and response
Data on treatments for catatonia in genetic conditions were available for 92 individuals. Of the 57 relevant studies, there were 3 cohort studies, 15 case series, and 39 case reports.
Benzodiazepines were administered to 64 individuals, of whom 18 (28%) had full remission, 27 (42%) partial remission, 11 (17%) no response, 1 (2%) clinical deterioration, 2 (2%) cessation due to adverse effects, and 5 (8%) an unspecified outcome. Antipsychotic medications were administered to 47 individuals, of whom 11 (23%) had full remission, 11 (23%) partial remission, 13 (28%) no response, 5 (11%) clinical deterioration, 2 (4%) cessation due to adverse effects, and 5 (11%) an unspecified outcome. ECT was administered to 43 individuals, of whom 26 (60%) had full remission, 10 (23%) partial remission, 4 (9%) no response, 2 (5%) clinical deterioration, 1 (2%) cessation due to adverse effects, and none with an unspecified outcome.
Discussion
To our knowledge, this paper provides the first comprehensive review of genetic abnormalities associated with catatonia. We identified three specific genetic conditions that may be associated with an increased prevalence of catatonia: Down’s syndrome, DiGeorge syndrome (22q11.2 DS), and PMS. We identified 44 other genetic abnormalities in which cases of catatonia have been described, representing a heterogeneous range of genetic abnormalities. The major focus of genetic association studies in catatonia has centered on identifying candidate genes for periodic catatonia at both 22q13 and 15q15 given early and influential studies by Stober et al. (Reference Stöber, Meyer, Nanda, Wienker, Saar, Jatzke, Schmid, Lesch and Beckmann2000, Reference Stöber, Seelow, Rüschendorf, Ekici, Beckmann and Reis2002). The increased prevalence of catatonia in PMS further implicates 22q13 as an area of interest; however, there is no replicated evidence in support of specific candidate genes at this location. It remains to be seen whether the genetic variants associated with catatonic schizophrenia will be the same as catatonia of all causes. The first GWAS of catatonia was recently conducted by Wilson et al. (Reference Wilson, Sealock, Straub, Raman, Kipp, Dittus, Heckers, Ely and Davis2024), with no SNPs reaching a genome wide level of significance. The sample size was small, however, and associations were revealed between catatonia and polygenic scores for anxiety, bipolar disorder, and schizophrenia.
In our population, the course of illness was complicated by more than one episode of catatonia in 28.1% of the population, similar to 25.1% in a hospitalized catatonia cohort (Rogers et al., Reference Rogers, Pollak, Begum, Griffin, Carter, Pritchard, Broadbent, Kolliakou, Ke, Stewart, Patel, Bomford, Amad, Zandi, Lewis, Nicholson and David2023). Reporting of ethnicity was limited; however, 65% of reported cases were of Asian ethnicity. While clinicians should remain mindful of cultural influences on catatonia, this result should be interpreted with caution given only one large Chinese study reported Asian ethnicity (Yang et al., Reference Yang, Li, Yang, Xiao, Wang, Ding, Zhao, Song, Yue, Zhang, Zhang and Lv2013).
Catatonia phenomenology and treatment in genetic abnormalities
We did not identify a profile of catatonic features that can clearly distinguish a genetic etiology across the lifespan. Stupor and mutism were the most frequently reported symptoms, in keeping with large catatonia cohorts not specifically looking at patients with genetic abnormalities (Dawkins et al., Reference Dawkins, Cruden-Smith, Carter, Amad, Zandi, Lewis, David and Rogers2022). There were no clear differences in treatment response between cohorts with catatonia with and without specified genetic abnormalities. ECT response rates in our genetic populations (60% full response, 23% partial remission, 5% no response) are broadly reflected in all causes of catatonia (Luchini et al., Reference Luchini, Medda, Mariani, Mauri, Toni and Perugi2015), as were benzodiazepine responses (Pelzer, Van Der Heijden, & Den Boer, Reference Pelzer, Van Der Heijden and Den Boer2018). These results indicate that catatonia presenting in patients with genetic abnormalities is not unique in its phenomenology or treatment. It is however important to recognize neurodevelopmental groups within our cohort may show diverging trends not explored in this paper.
Psychosocial precipitants of catatonia
The role of psychosocial precipitants in catatonia is widely described in the literature; catatonia has been conceptualized as a response to fear (Moskowitz, Reference Moskowitz2004). As with mental illness broadly, significant emotional trauma (Miles et al., Reference Miles, Takahashi, Muckerman, Nowell and Ithman2019), deprivation, and abuse have been identified as risk factors for catatonia (Dhossche, Ross, & Stoppelbein, Reference Dhossche, Ross and Stoppelbein2012). Many case reports in this review described psychosocial precipitants prior to the onset of catatonia. Termini et al. (Reference Termini, Anand, Hickox, Richter and Smith2023) described an adolescent female with a 22q11.2 deletion, with memory difficulties, reduced verbal output and oral intake, psychomotor slowing, along with blank staring periods following a traumatic occurrence with an intimate partner. Similarly, Torr and D’Abrera (Reference Torr and D’Abrera2014) reported a case of catatonia in Down’s syndrome triggered by the sudden deaths of two friends and the departure of a favorite staff member. Holm (Reference Holm2014) described a patient with a C9orf72 repeat expansion with cognitive decline following the loss of a close friend and a suicide attempt 6 months prior to presentation. Severe regression occurred for a patient after an untoward incident at school and for another patient after her father’s unexpected death (Miles et al., Reference Miles, Takahashi, Muckerman, Nowell and Ithman2019). Patients with genetic syndromes associated with learning difficulties may therefore be particularly vulnerable to developing catatonia as a stress response. Inequalities and discrimination often experienced by these populations can lead to increased social isolation and unmet mental health needs, increasing the vulnerability. There is evidence in the literature of a higher incidence of catatonia in Black individuals (Rogers et al., Reference Rogers, Pollak, Begum, Griffin, Carter, Pritchard, Broadbent, Kolliakou, Ke, Stewart, Patel, Bomford, Amad, Zandi, Lewis, Nicholson and David2023) and Asian individuals (Mustafa et al., Reference Mustafa, Bassim, Abdel Meguid, Sultan and Al Dardiry2012). This, as seen in our review, could prompt a consideration of exploring social disadvantages related to ethnicity or migration status.
Diagnosis
There are challenges to timely diagnosis and treatment of catatonia (Moyal et al., Reference Moyal, Plaze, Baruchet, Attali, Cravero, Raffin, Consoli, Cohen, Haroche and Chaumette2022; Stowe et al., Reference Stowe, Tyson, Hrynchak, White, Lang, Bailey, Macleod and Honer2014), including considerable heterogeneity in its presentation (Faedda et al., Reference Faedda, Wachtel, Higgins and Shprintzen2015). Individuals with neurodevelopmental disorders may show catatonia-like behavior, like increased slowness and difficulty initiating movement (Wing & Shah, Reference Wing and Shah2000), potentially creating difficulty recognizing catatonia in these patients. This is reflected in clinical data showing catatonia diagnosis is delayed by around 16 days in those with neurodevelopmental disorders (Zappia et al., Reference Zappia, Shillington, Fosdick, Erickson, Lamy and Dominick2024). The validity of rating scales has also been questioned in neurodevelopmental disorders. Some catatonic signs including mutism, grimacing, and agitation may differ in those with an intellectual disability compared to those without (Breckpot et al., Reference Breckpot, Vercruyssen, Weyts, Vandevoort, D’Haenens, Van Buggenhout, Leempoels, Brischoux-Boucher, Van Maldergem, Renieri, Mencarelli, D’Angelo, Mericq, Hoffer, Tauber, Molinas, Castiglioni, Brison, Vermeesch and Vogels2016). The Bush Francis Catatonia Rating Scale (BFCRS) does not account for regression symptoms in genetic syndrome-associated catatonia, which may lead to underestimated severity (Moyal et al., Reference Moyal, Plaze, Baruchet, Attali, Cravero, Raffin, Consoli, Cohen, Haroche and Chaumette2022). This may explain why improvement observed by families and medical teams often exceeds BFCRS measurements (Moyal et al., Reference Moyal, Plaze, Baruchet, Attali, Cravero, Raffin, Consoli, Cohen, Haroche and Chaumette2022). The BFCRS also lacks pediatric and neurodevelopmental-specific signs. Another challenge to prompt diagnosis could be diagnostic overshadowing or therapeutic blindness (Torr & D’Abrera, Reference Torr and D’Abrera2014), such as poorer recognition of depression in individuals with catatonia and ASD.
Catatonia is a syndrome exhibiting alterations in motor, behavioral, and vocal signs occurring in the context of medical, neurologic, and psychiatric disorders (Mazzone, Postorino, Valeri, & Vicari, Reference Mazzone, Postorino, Valeri and Vicari2014) The most common features are immobility, waxy flexibility, stupor, mutism, negativism, echolalia, echopraxia, peculiarities of voluntary movement, and rigidity (Medda et al., Reference Medda, Toni, Luchini, Giorgi Mariani, Mauri and Perugi2015). Despite substantial overlap between the DSM-5 and ICD-11 diagnostic criteria for catatonia, marked heterogeneity of a diagnosis of catatonia remains. In DSM-5, any combination of a minimum of 3 of 12 catatonic signs is used to diagnose catatonia, and catatonia has been made a specifier for 10 disorders, including schizophrenia, mood disorders, and general medical conditions. The four catatonia diagnoses in ICD-11 are catatonia associated with another mental disorder, catatonia induced by substances or medications, secondary catatonia syndrome, and catatonia unspecified.
Treatment
The most commonly used first-line treatments for catatonia in both children and adults are benzodiazepines and ECT, with this also observed in our review. In addition to GABA-A positive allosteric modulators and antipsychotics, NMDAR antagonists such as dextromethorphan/quinidine (Miles et al., Reference Miles, Takahashi, Muckerman, Nowell and Ithman2019) and mood stabilizers like lithium were used, especially when catatonia co-occurred with atypical bipolar affective disorder (Breckpot et al., Reference Breckpot, Vercruyssen, Weyts, Vandevoort, D’Haenens, Van Buggenhout, Leempoels, Brischoux-Boucher, Van Maldergem, Renieri, Mencarelli, D’Angelo, Mericq, Hoffer, Tauber, Molinas, Castiglioni, Brison, Vermeesch and Vogels2016; Verhoeven et al., Reference Verhoeven, Egger and De Leeuw2020). Individuals with copy number variants may respond differently to standard psychiatric treatment and when making treatment decisions for them, it is crucial to take into account the impact of multi-morbidity (Chawner, Watson, & Owen, Reference Chawner, Watson and Owen2021). Chronic catatonia when signs were not identified early was associated with a poor response to standard treatments like lorazepam and ECT (Moyal et al., Reference Moyal, Plaze, Baruchet, Attali, Cravero, Raffin, Consoli, Cohen, Haroche and Chaumette2022). In PMS benzodiazepines, while reducing catatonic signs, were sometimes observed to increase impulsivity, psychomotor arousal, confusion, and insomnia, possibly limiting their use in such cases (Mustafa et al., Reference Mustafa, Bassim, Abdel Meguid, Sultan and Al Dardiry2012). Unique treatment options tried included successful use of tDCS in 4 patients with PMS (Moyal et al., Reference Moyal, Plaze, Baruchet, Attali, Cravero, Raffin, Consoli, Cohen, Haroche and Chaumette2022), immunoadsorption in a case of Aicardi-Goutières syndrome (Ayrolles et al., Reference Ayrolles, Ellul, Renaldo, Boespflug-Tanguy, Delorme, Drunat, Elmaleh-Bergès, Kwon, Rozenberg, Bondet, Duffy, Crow and Melki2020), and levodopa in a case of Baraitser Winter syndrome (Diab et al., Reference Diab, Morin, Hery, Barbier, Cottin, Jobic and Tir2022). One study also reported the use of psychosocial approaches to augment treatment response; for example, Miles et al. (Reference Miles, Takahashi, Muckerman, Nowell and Ithman2019) found patients with Down’s syndrome improved more rapidly when they interacted with others during treatment, while withdrawal worsened when they were isolated.
Clinical course
There is evidence that patients may not return to their baseline level of functioning even after improvement in catatonia signs (Faedda et al., Reference Faedda, Wachtel, Higgins and Shprintzen2015). The re-emergence of signs of catatonia after cessation of therapy (Cowan et al., Reference Cowan, Lea, Flynn-Walden, Clothier and Grooms2022), breakthrough catatonia after ECT (Culleton et al., Reference Culleton, Hathaway, Poor, Wang and Holmes2022; Poser & Trutia, Reference Poser and Trutia2015), and protracted course of catatonia (Ghaziuddin et al., Reference Ghaziuddin, Miles and Nassiri2015) requiring maintenance ECT (Kohlenberg et al., Reference Kohlenberg, Trelles, McLarney, Betancur, Thurm and Kolevzon2020; Minamisawa et al., Reference Minamisawa, Sato, Saito, Takeuchi, Miyazaki, Odaka, Yamamoto, Oyama, Watanabe, Takeshita and Takahashi2023; Torr & D’Abrera, Reference Torr and D’Abrera2014) has been well described. In comparison to patients with psychiatric and medical disorders, catatonia in a Down’s syndrome cohort demonstrated a chronic course, requiring ongoing treatment to maintain recovery (Miles et al., Reference Miles, Takahashi, Muckerman, Nowell and Ithman2019).
Limitations
This work is limited by the quality of the data used which comprised mostly case reports. This study design was vulnerable to incomplete reporting of case histories and limited follow-up for key outcomes. Reporting of genetic testing methods was also incomplete across all study designs. Where methodology was reported, genetic testing was heterogeneous, and limited by the panels or markers included; potentially leading to oversimplified conclusions.
Nearly all of the association studies used pedigrees with periodic catatonia. Periodic catatonia is a very rare diagnosis with an estimated lifetime population prevalence of 0.001% (Stöber, Meyer, et al., Reference Stöber, Meyer, Nanda, Wienker, Saar, Jatzke, Schmid, Lesch and Beckmann2000). It is a type of catatonia in which signs are present episodically followed by periods of complete remission. It has been suggested that discrepancies between association studies could possibly be attributed to the use of the Leonhard (Reference Leonhard and Beckmann1999) system, which does not align with the ICD-10 diagnostic criteria and may not be synonymous with ‘catatonic schizophrenia’ (Selch et al., Reference Selch, Strobel, Haderlein, Meyer, Jacob, Schmitt, Lesch and Reif2007).
It is important to acknowledge the heterogeneous presentations of catatonia described in this paper. There is significant diagnostic heterogeneity because of the combinations of possible symptoms for diagnosing catatonia. Therefore, it is recommended that researchers report catatonia at the level of individual clinical signs in publications. This would enable researchers to test for genetic associations and treatment responses at the symptom level, improving the understanding of this heterogeneous condition. The diagnostic criteria for catatonia used in this paper do overlap considerably (Oldham, Reference Oldham2024) providing confidence that the clinical presentations do reflect a distinct entity. However, there remains the possibility that the range of genetic abnormalities found reflects distinct conditions, falsely diagnosed as catatonia. This represents pervasive diagnostic issue across complex phenotypes, including other neuropsychiatric disorders.
Future research
Our results point to a potential role of psychosocial triggers for catatonia, particularly in those with genetic syndromes. Future research should prioritize investigating gene–environment interactions in catatonia to ultimately guide targeted prevention and personalized treatment strategies for this complex neuropsychiatric condition. Use of heterogeneous genetic testing modalities limits the conclusions that can be drawn about genetic associations. Future research should focus on systematically genotyping large cohorts of patients with catatonia; particularly in populations with diversity in underlying condition and genetic ancestry.
Conclusions
Clinicians treating catatonia should be alert to the genetic disorders conferring clinical vulnerability. While specific syndromes such as Down’s syndrome, DiGeorge syndrome, and PMS have been frequently associated, our data show catatonia can occur in a wide range of genetic abnormalities. Catatonia often co-occurs with psychiatric conditions, but it is unclear if the same variants in the genome that predispose to psychiatric or intellectual disorders can also precipitate catatonia. Therefore, genetic research in catatonia patients may help uncover new therapeutic targets for this disorder (Breckpot et al., Reference Breckpot, Vercruyssen, Weyts, Vandevoort, D’Haenens, Van Buggenhout, Leempoels, Brischoux-Boucher, Van Maldergem, Renieri, Mencarelli, D’Angelo, Mericq, Hoffer, Tauber, Molinas, Castiglioni, Brison, Vermeesch and Vogels2016).
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1017/S0033291725100536
Data availability statement
Data available on reasonable request to the corresponding author.
Funding statement
CJW is supported by an NIHR Academic Clinical Fellowship (ACF-2022-17-009). JBF is supported by an NIHR Academic Clinical Fellowship (ACF-2023-13-016). JB is supported by NIHR Academic Clinical Fellowship (ACF-2024-17-014), IC is supported by NIHR Academic Clinical Fellowship (ACF-2024-19-014). MR is supported by the UK Academy of Medical Sciences and the Brain and Behaviour Research Foundation. KR, AD, BC, PS, and ER have no funding to disclose. Wellcome Trust (JPR; 220659/Z/20/Z).
Competing interests
Dr Rogers reports research funding from Wellcome and NIHR; royalties from Taylor & Francis; payment for reviewing from Johns Hopkins University Press; and speaker fees from the Alberta Psychiatric Association, Grey Nuns Hospital (Edmonton), Informed Research & Training Ltd., North East London NHS Foundation Trust, and Vanderbilt University Medical Center. He is a Council member for the British Association for Psychopharmacology, a member of the Medical Advisory Board of the Catatonia Foundation, and an Advisor to the Global Neuropsychiatry Group. He conducts expert witness work.
Dr Santosh is on the advisory board of Acadia Pharmaceuticals. PS was a past principal investigator (PI) on the Sarizotan (protocol number: Sarizotan/001/II/2015: ClinicalTrials.gov identifier: NCT02790034) and the ANAVEX Life Sciences Corp. (protocol number: ANVEX2–73-RS-003) clinical trial for Rett syndrome (RTT). PS is a co-inventor of the HealthTrackerTM and is the Chief Executive Officer of and shareholder in HealthTracker Ltd.
All other authors report no biomedical financial interest or potential conflicts of interest.







 Open access
Open access