


Introduction
Rheumatoid arthritis (RA) is an autoimmune disorder characterized by chronic joint inflammation, hyperplastic synovial pannus formation, and joint and bone destruction (Ref. Reference You, Koh and Leng1). Globally, RA impacts about 0.5% of the population, exhibiting a higher incidence in women than in men (Ref. Reference Smith and Berman2). Although targeted therapies like disease-modifying anti-rheumatic drugs, biologics, steroids and anti-inflammatory drugs have improved outcomes in RA patients, a considerable population of patients still exhibit persistent nonresponse (Refs. Reference Smolen, Landewé and Bergstra3, Reference Fraenkel, Bathon and England4, Reference Alivernini, Firestein and McInnes5, Reference Aletaha and Smolen6, Reference Nagy, Roodenrijs and Welsing7, Reference Svensson, Zoccheddu and Yang8). The variation in treatment responses indicates that RA involves diverse underlying mechanisms, despite the similarity in clinical symptoms among patients. This highlights the importance of identifying novel therapeutic targets. Recent studies have emphasized the critical role of mesenchymal-cell-derived cells, particularly fibroblast-like synoviocytes (FLS), in both initiating and perpetuating the complex processes in RA. Thus, developing strategies that specifically target FLS can offer new perspectives and significantly improve the treatment of RA (Refs. Reference Dakin, Coles and Sherlock9, Reference Nygaard and Firestein10, Reference Noss and Brenner11, Reference Pradhan, Krishnamurty and Fletcher12, Reference Wei, Nguyen and Brenner13, Reference Bartok and Firestein14).
The composition of synovial tissue in RA primarily consists of cells, including macrophage-like cells (MLCs), T lymphocytes, myeloid cells and FLS (Refs. Reference Zhang, Wei and Slowikowski15, Reference Floudas, Smith and Tynan16). FLS are crucial cell types lining articular joints, reflecting the inflammatory state and joint destruction in RA (Ref. Reference Carmona-Rivera, Carlucci and Moore17). The pathogenesis of RA begins with the abnormal activation of FLS, which proliferate excessively and resist apoptosis, leading to the formation of a hyperplastic synovial pannus. The pannus invades and erodes adjacent cartilage and bone, causing the characteristic joint damage observed in RA. FLS in RA also secrete pro-inflammatory cytokines, chemokines and matrix metalloproteinases (MMPs), which further exacerbate joint inflammation and degradation (Refs. Reference Croft, Campos and Jansen18, Reference Smolen, Aletaha and Barton19, Reference Firestein and McInnes20). Consequently, various aspects of the life cycle and activation of RA-FLS can be targeted, including their generation and proliferation, migration, direct destructive functions, and interactions with leukocytes (Ref. Reference Németh, Nagy and Pap21). Advances in molecular biology, immunology and computational biology have improved our understanding of the heterogeneous nature of FLS and their interactions with immune cells within the synovial environment of the joint. In this review, we highlight the role of FLS in promoting pannus formation and investigate the molecular mechanisms that regulate the balance between resident FLS and joint pathogenesis. We also emphasize the potential of targeting FLS and their signalling pathways, such as Janus kinase /signal transducer and activator of transcription (JAK/STAT), mitogen-activated protein kinase (MAPK), nuclear factor kappa B (NF-ĸB), Notch and interleukin-1 receptor-associated kinase 4 (IRAK4), as therapeutic strategies. These approaches hold promise for developing more effective treatments that could potentially halt or reverse the progression of RA. Moreover, we consider the fields of immunometabolism and computational biology, discussing their potential for therapeutic interventions targeting FLS. These insights may open up new avenues for developing FLS-targeted strategies for treating RA patients.
The physiological role of FLS
A healthy diarthrodial joint is lined by the synovium, a thin membrane composed of soft tissue. This structure typically consists of a few cell layers and includes type A synoviocytes (macrophage-like synoviocytes) and type B synoviocytes (FLS). FLS, as specialized mesenchymal cells, are essential in maintaining the structure of the synovium, featuring a substantial amount of rough endoplasmic reticulum. The synovium forms a protective membrane at the edges of joints, providing lubrication and nourishment to the cartilage through the production of synovial fluid (Ref. Reference Smith22), which is crucial for maintaining joint integrity (Ref. Reference Nygaard and Firestein10). Additionally, FLS are instrumental in regulating the synovial fluid and the extracellular matrix (ECM) of the joint lining, critical for maintaining the structural and functional integrity of diarthrodial joints. They actively produce various matrix components such as collagen, tenascin, laminin and proteoglycans, as well as enzymes that regulate ECM degradation, including MMPs, cathepsins and proteases (Ref. Reference Alivernini, Firestein and McInnes5).
FLS also serve as vigilant immune sentinels that are capable of producing cytokines, small-molecule mediators and proteases, thereby forming the synovial intima (Ref. Reference Carmona-Rivera, Carlucci and Moore17). The observation that inflammation is suppressed in mice lacking the FLS scaffold further highlights their immunomodulatory effects (Ref. Reference Noss and Brenner11). Beyond their role in producing pro-inflammatory mediators, FLS contribute to a synovial matrix that encompasses macrophages and other leukocytes, thus facilitating the activation of these cells.
The pathological transformations of FLS in RA
In RA, the infiltrating synovium primarily consists of FLS, comparable to tumour cells (Refs. Reference Nygaard and Firestein10, Reference Bartok and Firestein14). Studies indicated that an increase in FLS contributes to inflammation, hyperplasia and aggressiveness in the synovial lining, triggered by exposure to platelet-derived growth factor (PDGF), tumour necrosis factor (TNF) or interleukin (IL)-1. This leads to the formation of a destructive tissue known as pannus (Refs. Reference Beckmann, Römer-Hillmann and Krause23, Reference Wu, Ma and Yang24), which damages the cartilage and non-osseous structural elements of the joint microenvironment in RA (Refs. Reference Alivernini, MacDonald and Elmesmari25, Reference Buckley26, Reference Buckley and McGettrick27) (see Figure 1). Targeting FLS was considered a critical therapeutic approach in managing RA, focussing on reducing inflammation and halting disease progression (Refs. Reference Noss and Brenner11, Reference Muller-Ladner, Pap and Gay28).
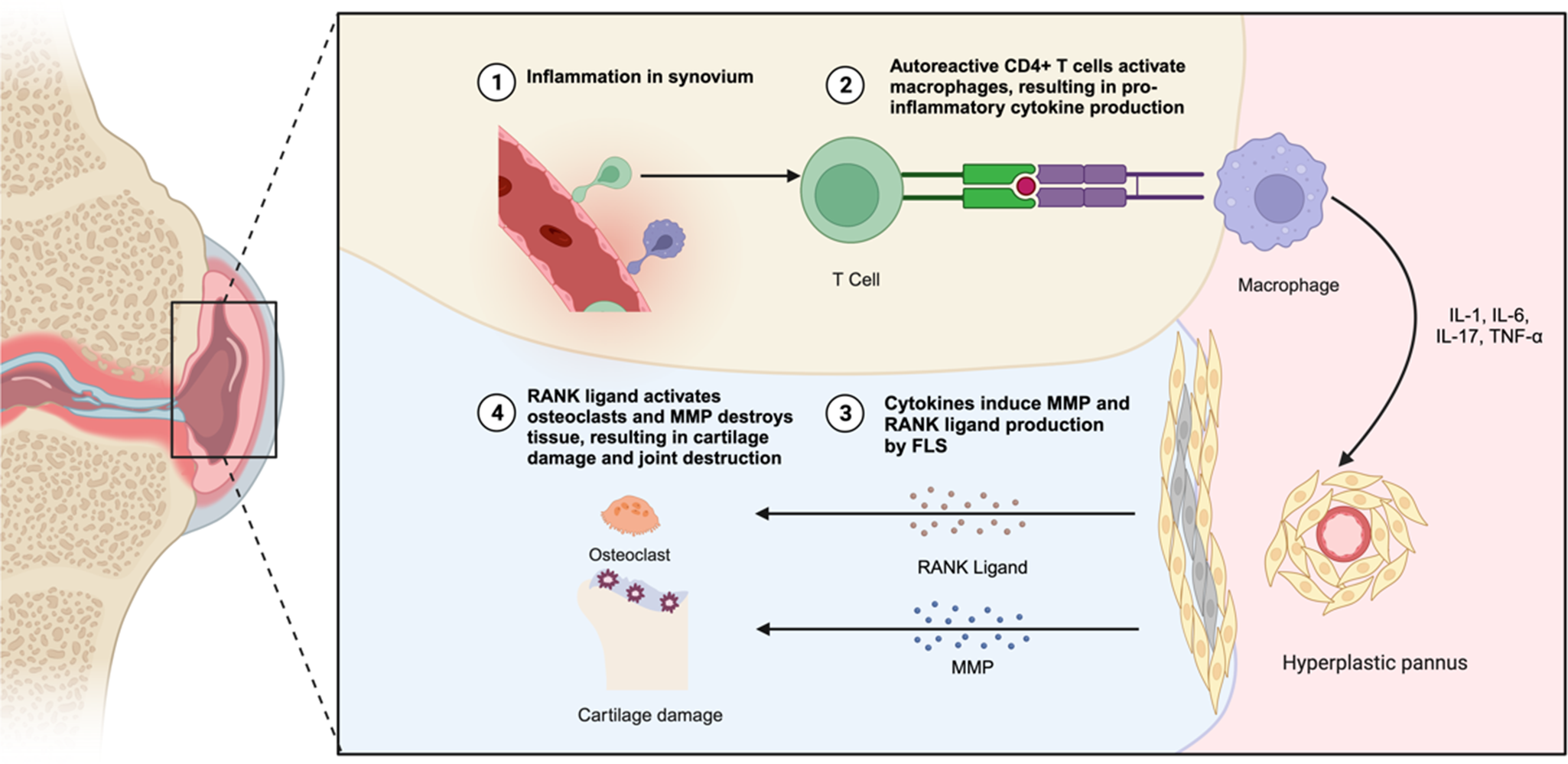
Figure 1. The pathological changes in the RA synovial joint. In RA, the synovial lining undergoes hyperplasia, forming an invasive pannus that contributes to joint destruction. FLS play a central role in this process by producing pro-inflammatory cytokines, matrix metalloproteinases (MMPs) and chemokines that drive chronic inflammation and cartilage degradation. Additionally, FLS interact with immune cells such as macrophages, which secrete TNF-α and IL-1β, further amplifying inflammatory signalling, and T cells, which activate FLS via IL-17 and other cytokines. This crosstalk promotes the recruitment and activation of osteoclast precursors, enhancing bone erosion while simultaneously inhibiting osteoblast-mediated bone repair. The combined effects of these pathological interactions lead to irreversible joint damage and progressive disability in RA patients.
The impact of FLS proliferation and pannus formation in RA
FLS proliferation significantly contributes to RA pathogenesis through excessive proliferation in the synovial lining and sublining. Several factors drive their proliferation, including resistance to apoptosis, enhanced autophagy, growth factors, inflammatory cytokines and stress responses with the endoplasmic reticulum stress (ERS) (Refs. Reference Bottini and Firestein29, Reference Lafyatis, Remmers and Roberts30, Reference Korb, Pavenstadt and Pap31, Reference Kato, Ospelt and Gay32, Reference Shin, Han and Kim33). Additionally, the differentiation and migration of mesenchymal stem cells into the synovium further promote FLS accumulation, enhancing synovial tissue thickening (hyperplasia) (Refs. Reference Steenvoorden, Tolboom and van der Pluijm34, Reference Marinova-Mutafchieva, Williams and Funa35). Recent studies have identified pre-inflammatory mesenchymal (PRIME) cells, which infiltrate the synovium during RA flare-ups. These PRIME cells interact with endothelial cells, subsequently differentiating into sublining FLS subsets, thus contributing to synovial expansion and disease progression (Refs. Reference Chu36, Reference Orange, Yao and Sawicka37). The proliferation and invasive characteristics of FLS also drive the formation of pannus, an aggressive fibrovascular tissue that contributes directly to joint damage in RA. Within pannus tissue, FLS exhibit tumour-like properties, including increased invasiveness, migration and resistance to normal growth inhibition, enabling their spread and infiltration into joint structures (Refs. Reference You, Koh and Leng1, Reference Bottini and Firestein29) These pannus-resident FLS, influenced by inflammatory mediators such as galectin-3, produce large amounts of matrix-degrading enzymes, cytokines and chemokines, accelerating immune cell infiltration, cartilage destruction and joint erosion (Refs. Reference Buckley, Ospelt and Gay38, Reference Mendez-Huergo, Hockl and Stupirski39).
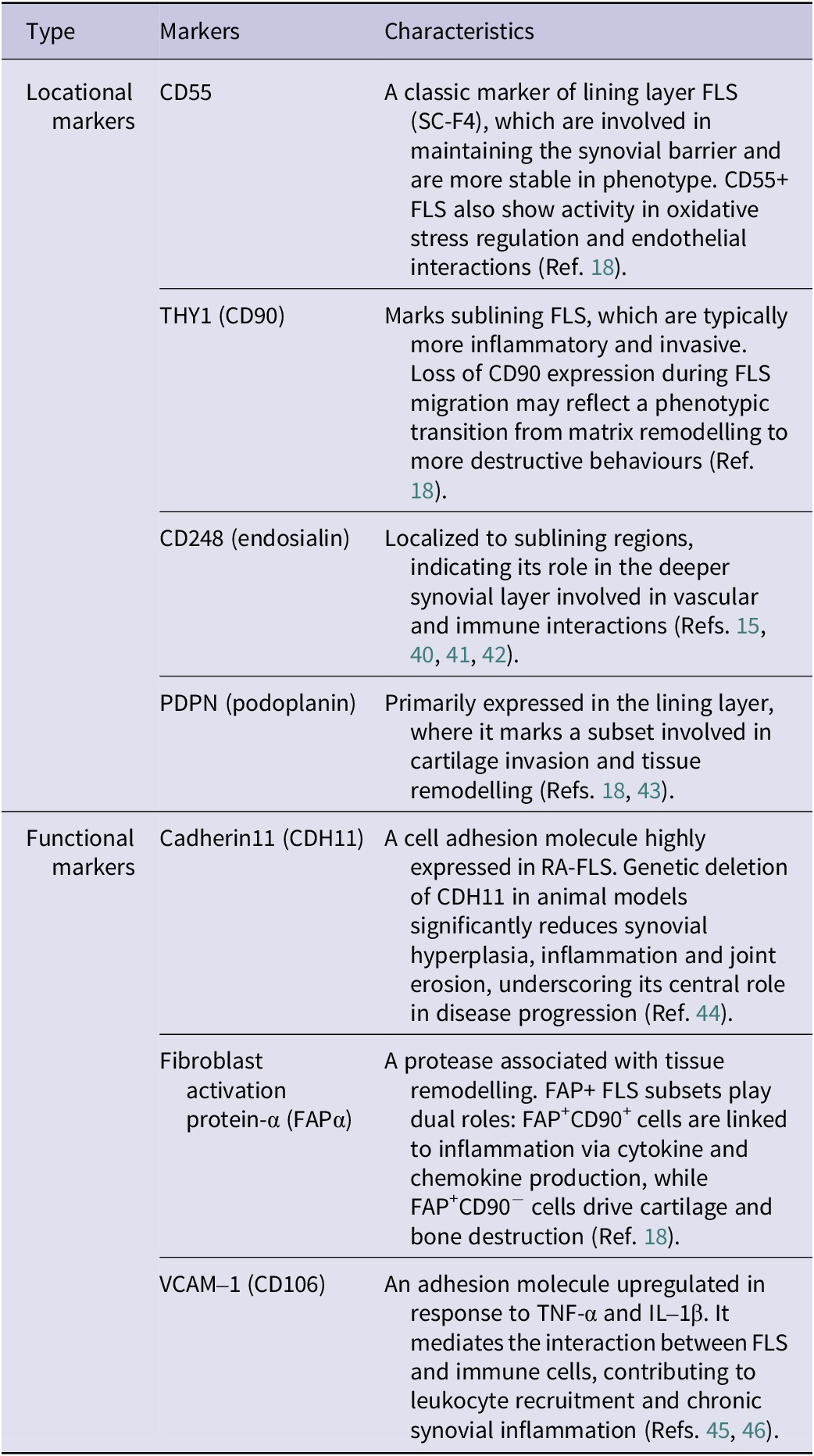
Within the synovium, FLS exhibit marked heterogeneity, which can be classified based on their functional roles and anatomical localization (see Table 1). Beyond surface marker expression, single-cell RNA sequencing (scRNA-seq) has offered deeper insight into the dynamic phenotypes of FLS subsets in RA. ScRNA-seq studies from both human RA patients and murine models have revealed a conserved landscape of FLS subsets implicated in disease pathogenesis. In mice, distinct fibroblast populations localize to the synovial lining and sublining regions, with inflammatory and matrix-remodelling subsets expanding during arthritis progression (Ref. Reference Croft, Campos and Jansen18). Likewise, human RA synovium harbours transcriptionally analogous populations, including immunofibroblasts expressing human leukocyte antigen-DR alpha (HLA-DRA), IL-6, and destructive fibroblasts marked by MMP-3, MMP-9 and FAP (Ref. Reference Zhang, Wei and Slowikowski15). Cross-species comparisons confirm the conservation of these pathogenic phenotypes, indicating that murine models effectively recapitulate human FLS heterogeneity. Notably, therapeutic targeting of specific subsets – such as FAP+ fibroblasts – in mice reduces inflammation and joint damage, emphasizing the translational potential of subset-specific strategies in human RA (Ref. Reference Dorst, Rijpkema and Boss47). Overall, identifying these functional and locational markers provides valuable insights into RA pathogenesis and supports targeted therapeutic strategies aimed at reducing inflammation and preserving joint integrity.
Table 1. The key markers expressed by FLS
The role of macrophages and FLS interactions in RA
Under normal conditions, macrophages are key cellular components of the synovium. Using scRNA-seq and immunometabolism analyses, various subpopulations of macrophages with distinct homoeostatic, regulatory and inflammatory functions have been identified within the synovium (Ref. Reference Alivernini, MacDonald and Elmesmari25). In RA, the interaction between FLS and macrophages plays a critical role in both the initiation of synovial inflammation and subsequent joint destruction.
CX3CR1+ macrophages, which form a protective barrier layer alongside fibroblasts in the synovial tissue, comprise 40% of the macrophage population under resting conditions. Their removal in experimental arthritis models in mice leads to the infiltration of cells derived from monocytes and neutrophils, damaging the synovial barrier. These findings highlight the critical role of CX3CR1+ macrophages as a primary immune-regulatory checkpoint in controlling synovial inflammation. Dysfunction of this checkpoint results in chronic joint inflammation. When CX3CR1+ macrophages are removed from the synovium in mice with experimental arthritis, an influx of neutrophils and monocyte-derived cells into the synovium occurs, causing damage to the synovial barrier (Ref. Reference Culemann, Gruneboom and Nicolas-Avila48). Recently, Alivernini et al. observed that compared to patients with active RA, healthy synovium and patients in remission from RA had higher numbers of MERTK+ TREM2high and LYVE1+ macrophages. Synovial tissues with a lower number of these cellular subgroups were more prone to flare-ups (Ref. Reference Alivernini, MacDonald and Elmesmari25). MERTK+ macrophages are known to produce pro-resolving lipids and promote FLS repair, while MERTK− macrophages proliferate and release high levels of TNF-α and IL-6 during the active phase, leading to pathogenic FLS activation (Ref. Reference Alivernini, MacDonald and Elmesmari25). Thus, MERTK+ macrophages act as a crucial inhibitory mechanism to counteract pro-inflammatory FLS behaviours. In the absence of this checkpoint, pathogenic FLS subsets may develop with sustained activation.
FLS release chemokines such as CCL2, CCL5, CCL8, CXCL5 and CXCL10 in response to inflammatory stimuli, which lead to the recruitment of macrophages and monocytes (Ref. Reference Bartok and Firestein14). RA is driven by cytokine networks at the sites of inflammation (Refs. Reference Naylor, Filer and Buckley49, Reference Lee, Qiao and Grigoriev50, Reference Migita, Iwanaga and Izumi51). TNF-induced soluble factors from RA-FLS inhibit the expression of type I interferon-regulated genes in macrophages by downregulating JAK/STAT signalling (Ref. Reference Donlin, Jayatilleke and Giannopoulou52). Alongside pro-inflammatory factors, prostaglandins produced by RA-FLS induce macrophages into a state characterized by an increased production of pro-heparin-binding EGF-like growth factor (HBEGF) and pro-inflammatory genes (Ref. Reference Kuo, Ding and Cohn53). It is widely recognized that macrophages are the primary source of IL-1β and TNF, while FLS in the intimal lining are the main producers of IL-6 (Ref. Reference Alvaro-Gracia, Zvaifler and Firestein54). Additionally, FLS in the intimal lining secrete colony-stimulating factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage-colony stimulating factor (M-CSF) (Ref. Reference Firestein55). IL-1β/TNF-stimulated FLS upregulate GM-CSF production, further facilitating the recruitment and activation of macrophages. Therapies targeting CXCL10 and GM-CSF receptors have shown promising results in RA treatment (Refs. Reference Yellin, Paliienko and Balanescu56, Reference Burmester, McInnes and Kremer57).
Another aspect of the complex interaction between macrophages and FLS is the differentiation of macrophages into osteoclasts, specialized cells that absorb bone. Multinucleated osteoclasts perform extensive bone resorption at the apex of the pannus (Ref. Reference Yoshitomi58). A key factor in the differentiation of macrophages into osteoclasts is receptor activator of nuclear factor kappa B ligand (RANKL) (Refs. Reference Fuller, Wong and Fox59, Reference Lacey, Timms and Tan60). Upon activation, FLS can produce a significant amount of RANKL and M-CSF (Ref. Reference Pincus and Sokka61). Clinical trials involving anti-RANKL antibodies have shown significant efficacy in reducing bone loss associated with RA (Ref. Reference Cohen, Dore and Lane62). Moreover, RA-FLS inhibit the activation of osteoblasts by secreting Dicckopf-1, a regulator of the Wnt signalling pathway, thereby impeding the repair processes of bone erosions (Ref. Reference Diarra, Stolina and Polzer63).
The epigenic modification in RA-FLS
Emerging evidence underscores the significance of epigenetic modifications, including DNA methylation, SUMOylation, histone modifications and non-coding RNA (ncRNA) expression, in the pathogenesis of RA (Refs. Reference Nakano, Boyle and Firestein64, Reference Ai, Whitaker and Boyle65, Reference Ai, Laragione and Hammaker66). These epigenetic changes critically influence FLS behaviour, promoting inflammation, invasion and joint damage. Additionally, genetic mutations in TP53 (Ref. Reference Igarashi, Hashimoto and Tomita67) contribute further to apoptosis resistance, enhanced survival and invasiveness of RA-FLS. Furthermore, mitochondrial dysfunction induced by inflammatory signalling exacerbates oxidative stress and inflammation in the synovium (Refs. Reference Da Sylva, Connor and Mburu68, Reference Promila, Joshi and Khan69).
DNA methylation, a crucial epigenetic modification regulating gene expression, is increasingly recognized as a key driver of the aggressive and invasive behaviour of FLS in RA. RA-FLS display distinct genome-wide DNA methylation patterns compared to healthy synovial fibroblasts, characterized by widespread hypomethylation, particularly in promoter regions of genes involved in inflammation, matrix degradation and cellular adhesion. This altered methylation landscape facilitates aberrant gene expression, promoting synovial hyperplasia and joint destruction (Refs. Reference Nakano, Boyle and Firestein64, Reference Ai, Laragione and Hammaker66). This aberrant methylation pattern leads to the overexpression of pro-inflammatory cytokines and MMPs, thereby enhancing synovial inflammation and tissue invasion. One key target gene, phosphatase and tensin homolog (PTEN), known for its anti-inflammatory and anti-proliferative effects, is epigenetically silenced in RA-FLS through promoter hypermethylation. This modification is induced by pro-inflammatory stimuli such as TNF-α and contributes to the increased production of cytokines and chemokines, as well as enhanced FLS proliferation and migration (Refs. Reference Li, Xin and Jing70, Reference Li, Wu and Yan71). Additionally, the upregulation of DNA methyltransferases (DNMTs), particularly DNMT1, further contributes to maintaining this aberrant methylation landscape (Ref. Reference Li, Wu and Yan71). Integrative epigenomic and transcriptomic analyses have identified numerous hypomethylated, overexpressed genes in RA-FLS, including Huntingtin-interacting protein 1 (HIP1) and other regulators of cytoskeletal remodelling and cell migration, providing mechanistic insights into FLS invasiveness (Refs. Reference Li, Xin and Jing70, Reference Li, Wu and Yan71, Reference Ge, Frank-Bertoncelj and Klein72). Collectively, these findings highlight DNA methylation as a dynamic and functional contributor to the pathological activation of RA-FLS. Targeting specific epigenetic alterations, particularly those regulating key genes like PTEN and HIP1, may offer novel therapeutic strategies to limit joint damage and inflammation in RA.
Epigenetic modifications such as SUMOylation and histone modifications play integral roles in the pathological activation and invasive phenotype of FLS in RA. SUMOylation, a post-translational process involving the covalent attachment of small ubiquitin-like modifier (SUMO) proteins to lysine residues of target proteins, regulates their stability, localization and interactions (Ref. Reference Tharuka, Courelli and Chen73). In RA-FLS, components of the SUMOylation pathway, including the SUMO-activating enzyme SAE1/UBA2 and the conjugating enzyme UBC9, are significantly upregulated (Refs. Reference Dehnavi, Sadeghi and Johnston74, Reference Wang, Xiao and Lao75, Reference McHugh76, Reference Li, Li and Kou77). This enhanced SUMOylation promotes glycolytic reprogramming via modification of pyruvate kinase M2 (PKM2) and increases the secretion of inflammatory mediators such as vascular endothelial growth factor (VEGF)-A, MMP-3 and MMP-9, thereby driving FLS proliferation, migration and joint invasion. Importantly, SUMOylation and histone modifications are tightly linked processes; SUMOylation can directly modify histones or modulate histone-modifying enzymes, thereby influencing chromatin structure and gene expression (Ref. Reference Ryu and Hochstrasser78). A key regulatory axis in this system is the balance between SUMO ligases and deSUMOylating enzymes like SENP1. In RA-FLS, SENP1 is downregulated, contributing to excessive SUMOylation of transcription factors (TFs) and histones (Ref. Reference Sheng, Luo and Huang79). Notably, SUMOylation enhances histone H4 acetylation at the MMP-1 promoter, promoting its transcription and contributing to cartilage degradation. Restoration of SENP1 reverses these effects by reducing histone acetylation and MMP-1 expression (Ref. Reference Maciejewska-Rodrigues, Karouzakis and Strietholt80). Inflammatory cytokines such as TNF-α further amplify this epigenetic dysregulation by increasing histone deacetylase (HDAC) activity, which modifies chromatin structure and sustains pro-inflammatory gene expression (Ref. Reference Kawabata, Nishida and Takasugi81). This interplay creates a feedback loop that reinforces transcriptional programmes associated with inflammation and tissue invasion. Moreover, dysregulated SUMOylation intersects with key signalling pathways such as Wnt, further driving FLS proliferation and invasiveness (Ref. Reference Fan, Yang and Zheng82). A notable example is the overexpression of the histone methyltransferase enhancer of zeste homolog 2 (EZH2), which catalyses H3K27 trimethylation and represses secreted frizzled-related protein 1 (SFRP1) – an endogenous inhibitor of the Wnt pathway (Ref. Reference Trenkmann, Brock and Gay83). Suppression of SFRP1 by EZH2 promotes unchecked Wnt signalling, exacerbating RA synovial pathology. Collectively, these findings highlight how SUMOylation acts in concert with histone modifications – particularly acetylation and methylation – to reprogramme RA-FLS towards an aggressive, tissue-destructive phenotype. Targeting components of these pathways, such as SAE1/UBA2, UBC9, SENP1, HDACs and EZH2, offers promising therapeutic potential to reduce synovial inflammation and joint damage in RA.
ncRNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), also regulate RA-FLS behaviour by modulating gene expression at transcriptional and post-transcriptional levels (Refs. Reference Wijesinghe, Lindsay and Jones84, Reference Chang, Xu and Zhang85). miRNAs typically suppress target gene expression by binding to messenger RNAs (mRNAs), causing mRNA degradation or inhibiting translation. Researchers found that miR-221 levels were increased in the serum and synovial tissues of RA patients compared to healthy controls (Ref. Reference Yang and Yang86). When miR-221 was downregulated in FLS stimulated with lipopolysaccharide (LPS), there was a significant decrease in the expression of pro-inflammatory cytokines and chemokines. Additionally, the downregulation of miR-221 inhibited FLS migration and invasion, which was associated with the reduced expression of VEGF, MMP-3 and MMP-9. These findings suggest that miR-221 contributes to RA pathogenesis by promoting inflammation and joint destruction through the upregulation of MMPs and other pro-inflammatory mediators (Refs. Reference Yang and Yang86, Reference Pandis, Ospelt and Karagianni87). Conversely, lncRNAs regulate gene expression primarily via chromatin remodelling or recruiting transcriptional regulators. Homoeobox antisense intergenic RNA (HOTAIR), a lncRNA overexpressed in RA-FLS, can modulate the expression of specific MMPs such as MMP-2 and MMP-13 through epigenetic mechanisms. While increased HOTAIR typically suppresses certain MMPs, in various pathological contexts it paradoxically enhances cell invasiveness, illustrating context-dependent regulatory roles of lncRNAs (Refs. Reference Huh, Lee and Song88, Reference Gupta, Shah and Wang89).
Collectively, epigenetic modifications – DNA methylation, SUMOylation, histone acetylation and methylation, and ncRNA regulation – are central mechanisms governing the aggressive phenotype of RA-FLS. Improved understanding of these epigenetic pathways offers novel therapeutic avenues to mitigate RA-FLS-driven inflammation, invasiveness and joint destruction, ultimately improving patient outcomes.
Immunometabolism of FLS in RA
Discoveries in immunology have highlighted the importance of metabolic adaptations during the early stages of immune responses, now recognized as essential processes (Refs. Reference Febbraio, Hiscock and Sacchetti90, Reference Hotamisligil, Shargill and Spiegelman91, Reference Pearce, Walsh and Cejas92, Reference Wieman, Wofford and Rathmell93, Reference Lercher, Baazim and Bergthaler94). Increasing evidence has shown that metabolic changes affect stromal and immune cells (Refs. Reference Ghesquière, Wong and Kuchnio95, Reference Wang and Green96, Reference Pearce, Poffenberger and Chang97) and play a role in autoimmune diseases (Refs. Reference Yin, Choi and Xu98, Reference Yang, Matteson and Goronzy99, Reference McDonald, Deepak and Miguel100). Various factors within the cellular microenvironment activate key signalling pathways that influence cellular metabolism, supporting cell growth and survival (Refs. Reference Cairns, Harris and Mak101, Reference Vander Heiden, Cantley and Thompson102). Notably, metabolic disruption has been associated with RA (Ref. Reference Ahn, Kim and Hwang103). In RA, FLS exhibit a unique metabolic profile within the inflamed joint milieu. Characterized by a hypermetabolic phenotype, FLS undergo substantial metabolic shifts that augment their proliferative and invasive capacities (Refs. Reference Falconer, Murphy and Young104, Reference Weyand and Goronzy105, Reference Aghakhani, Zerrouk and Niarakis106).
Glucose metabolism of FLS in RA
As the first step of glucose metabolism, glycolysis often occurs in the cytoplasm of cells and produces only a small amount of total energy stored in adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NADP). Unlike normal cells, cancer cells tend to favour glycolysis to metabolize glucose over the more efficient oxidative phosphorylation, even in the presence of oxygen (the Warburg effect) (Ref. Reference Hsu and Sabatini107). Similar to cancer cells, RA-FLS also exhibit a Warburg-like metabolism, marked by increased glucose consumption and glycolytic flux (Ref. Reference Vander Heiden, Cantley and Thompson102). Garcia-Carbonell et al. (Ref. Reference Garcia-Carbonell, Divakaruni and Lodi108) have observed an increased expression of glucose transporter 1 (GLUT1) mRNA in the synovial lining cells in a model of inflammatory arthritis. Additionally, glucose uptake and glycolytic gene expression were increased in the stromal compartment of arthritic mouse joints, indicating significant alterations in glucose metabolism in RA-FLS. This study also has highlighted the effectiveness of 3-bromopyruvate (BrPA) in alleviating RA in a mouse model of inflammatory arthritis by targeting hexokinase-2 (HK2) through inhibiting glycolysis. Other glycolytic inhibitors, such as 2-deoxyglucose (2-DG) (Ref. Reference Abboud, Choi and Kanda109) and lonidamine (Ref. Reference Song, Lu and Fan110), have also been shown to inhibit glycolysis and regulate the inflammatory phenotype of RA-FLS, offering potential therapeutic approaches for managing RA.
Lactate metabolism of FLS in RA
Lactate production primarily occurs during conditions of increased aerobic glycolysis (Ref. Reference Rabinowitz and Enerbäck111). In the inflamed joint, there is a notable decrease in synovial fluid pH alongside an increase in lactate concentration, stemming from the intense cellular turnover within the synovium (Ref. Reference Pucino, Certo and Varricchi112). Monocarboxylate transporter 4 (MCT4) plays a role in transporting lactate out of tumour cells, supporting metastasis and angiogenesis (Refs. Reference Ullah, Davies and Halestrap113, Reference Pettersen, Ebbesen and Gieling114). The expression levels of MCT4 mRNA and protein were markedly increased in RA-FLS compared to OA-FLS (Ref. Reference Fujii, Kawahito and Nagahara115). Using specific siRNA to silence MCT4 has demonstrated a reduction in arthritis severity in mouse models of RA, underscoring MCT4 as a promising therapeutic target for RA (Ref. Reference Fujii, Kawahito and Nagahara115).
Lipid metabolism of FLS in RA
Although our understanding continues to evolve, it is acknowledged that lipids play a pivotal role in both fuelling adaptive immunity and dampening inflammation (Refs. Reference Ahn, Kim and Hwang103, Reference O’Neill, Kishton and Rathmell116). A notable finding was impaired mitochondrial fatty acid β-oxidation in FLS from individuals at risk of RA and RA patients, compared to healthy controls (Ref. Reference de, Semmelink and Denis117). This deficiency, associated with decreased gene expression in the β-oxidation pathway, suggests a lipid metabolic inflexibility that could contribute to the onset of RA. In addition, there was a marked increase in phospholipid levels, notably phosphocholine (PCho), in tumour cells (Ref. Reference Glunde, Bhujwalla and Ronen118). These increases in PCho were partially attributed to the increased activity of the enzyme choline kinase α (ChoKα), crucial for phosphatidylcholine (PtdCho) biosynthesis (Ref. Reference Lacal119). Guma et al. (Ref. Reference Guma, Sanchez-Lopez and Lodi120) have found that ChoKα was increased in RA synovium, further demonstrating that targeting ChoKα could be effective in reducing migration and promoting apoptosis in RA-FLS. The ChoKα inhibitor MN58b has proven to be effective in the reduction of harmful behaviours in FLS, such as migration and resistance to apoptosis, which are vital in the progression of RA (Ref. Reference Guma, Sanchez-Lopez and Lodi120).
Mitochondrial metabolism of FLS in RA
Mitochondria consume oxygen to generate ATP and produce metabolic intermediates of the tricarboxylic acid (TCA) cycle, participating in numerous metabolic pathways (Ref. Reference Kelly and O’Neill121). In RA patients’ synovial fluid, TCA intermediates like glutamate, citrate and succinate act as inflammation promoters (Ref. Reference Weyand and Goronzy105). Increased glutamate levels in arthritic joints enhance IL-6 release through the activation of glutamate receptors, contributing to arthritic pain (Ref. Reference Bonnet, Williams and Gilbert122). Research has indicated that glutaminase (GLS) 1, which catalyses the conversion of glutamine to glutamate, is upregulated in RA-FLS. Inhibiting GLS1 through siRNA transfection or with inhibitors can suppress the growth of RA-FLS (Ref. Reference Takahashi, Saegusa and Sendo123). Furthermore, the mitochondrial complex I inhibitor IACS-010759 disrupts RA-FLS metabolic rewiring by interfering with hypoxia-inducible factor (HIF) 1α and enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) regulation, reducing citrate or succinate accumulation (Ref. Reference Umar, Palasiewicz and Volin124). However, IACS-010759 showed inefficacy in a collagen-induced arthritis (CIA) preclinical model, cautioning its clinical application (Ref. Reference Umar, Palasiewicz and Volin124). Consequently, targeting mitochondrial metabolism, particularly the metabolic intermediates, offers a promising therapeutic approach for managing RA, potentially affecting both cell proliferation and the inflammatory response.
In conclusion, research has shown that the immune microenvironment in RA induces metabolic reprogramming in FLS, leading to dysfunction and imbalance in immune homoeostasis. Therefore, targeting this metabolic reprogramming could open new avenues for RA treatment.
Using computational biology in the characterization of RA-FLS
Bioinformatics is one of the most dynamic fields in biological research, with numerous studies utilizing computational platforms to delineate the developmental progress and tissue specification of various cell lines. A notable example included the work by Zhang et al. (Ref. Reference Zhang, Wang and Zhao125), who developed Taiji, an advanced algorithm for identifying key TFs essential for lineage-specific and stage-dependent tissue differentiation. This system-level strategy integrated transcriptomic and epigenomic data to accurately pinpoint essential TFs. Using the Taiji platform, Ainsworth et al. (Ref. Reference Ainsworth, Hammaker and Nygaard126) conducted a comprehensive analysis of primary RA-FLS cell lines, focussing on their distinct TF biology at the transcriptional level. Through their analysis, they identified that RA-FLS cell lines can be divided into two distinct groups, CL1 and CL2, based on their Personalized PageRank scores. This classification revealed significant differences in phenotypic traits and pathway activities between the groups. They identified cluster-specific TFs that played differential roles in the function of RA-FLS. Notably, retinoic acid receptor alpha (RARα) emerged as a crucial factor, exhibiting unique regulatory effects on TGFβ signalling pathways across the clusters. Further experimental validation confirmed the distinct roles of RARα within these clusters, particularly in its impact on gene expression, protein levels and cellular behaviours such as proliferation and invasion. These findings highlighted divergent TGFβ signalling and biological outcomes between the clusters, highlighting the molecular complexity of RA pathogenesis. This research not only biologically validated the predictions for the key subtype-specific TF, RARα, but also demonstrated the differential regulation of TGFβ signalling in the two subtypes, providing deeper insights into the variable clinical responses observed in RA treatments.
Various studies have employed different computational systems to explore the characteristics of RA-FLS. Aghakhani et al. (Ref. Reference Aghakhani, Soliman and Niarakis127) introduced a hybrid modelling approach to studying metabolic reprogramming in RA-FLS. Their model merged a qualitative regulatory network with a global metabolic network, using flux balance analysis to assess the impact of regulatory outcomes on metabolic flux distributions. It demonstrated how RA-FLS transition towards functioning as metabolic factories, potentially exacerbating RA pathology through increased energy and nutrient production, driven by HIF1 activation. Singh et al. (Ref. Reference Singh, Naldi and Soliman128) developed a comprehensive Boolean model to investigate FLS in RA. This model predicted drug synergies and identified potential new therapeutic targets by simulating various phenotypes such as inflammation and bone erosion. Additionally, it employed drug repurposing analysis to identify candidate drugs, thereby aiming to improve treatment strategies through a system-level understanding of RA pathophysiology. Ge et al. (Ref. Reference Ge, Frank-Bertoncelj and Klein72) provided a detailed genomic atlas of FLS, highlighting their role in the genetic predisposition to RA. By integrating DNA architecture, chromatin interactions and gene expression data, the study pinpointed genes and pathways that contribute to the heritability of RA, emphasizing the critical role of FLS in the disease’s pathogenesis. You et al. (Ref. Reference You, Yoo and Choi129) focussed on the molecular signatures that characterize the invasive behaviour of RA-FLS. The study used transcriptome profiling and network modelling to identify key regulators of FLS invasiveness, offering insights into the cellular mechanisms underlying RA pathogenesis and identifying potential therapeutic targets. Together, these studies improved our understanding of RA by covering various aspects, from metabolic changes and drug response predictions to genetic susceptibility and the invasive characteristics of FLS.
Signalling pathways in RA-FLS and prospects for therapeutic interventions
Several intracellular signalling pathways regulate the pathogenic behaviour of FLS, including the JAK/STAT, MAPK, NF-κB, Notch and IRAK4 signalling pathway. Given their crucial role, these signalling cascades represent promising therapeutic targets for RA treatment.
JAK/STAT signalling pathway
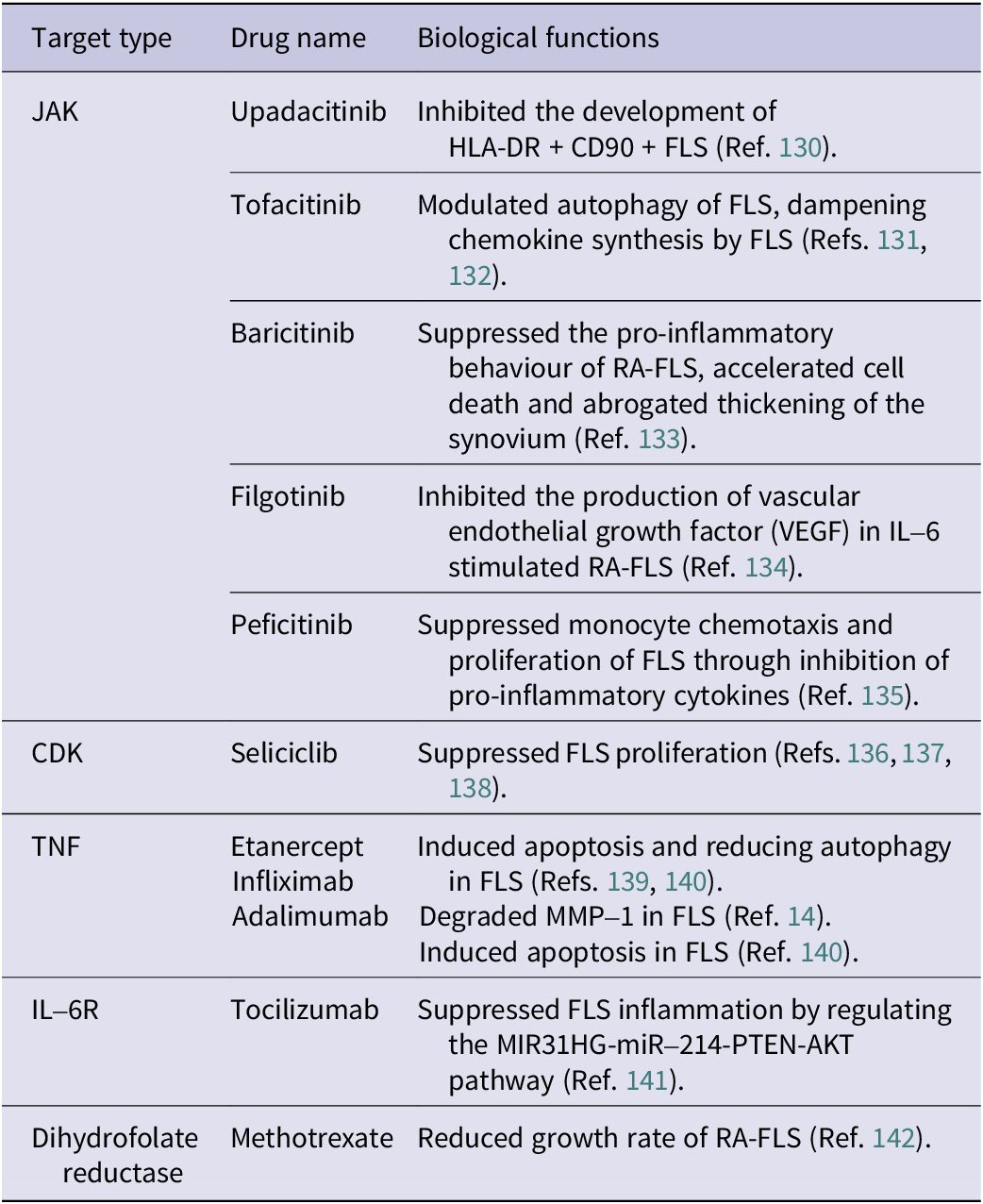
The JAK/STAT pathway plays a critical role in regulating the pathogenesis and progression of RA (Ref. Reference Malemud143). More than 50 cytokines are known to activate the JAK/STAT pathway through interactions with type I or type II cell-surface receptors, underscoring the importance of this signalling cascade in immune regulation and inflammation (Refs. Reference Morris, Kershaw and Babon144, Reference Gadina, Le and Schwartz145). The JAK family comprises four members: JAK1, JAK2, JAK3 and tyrosine kinase-2 (TYK2) (Ref. Reference Ghoreschi, Laurence and O’Shea146). These kinases transmit extracellular cytokine signals to the intracellular environment, initiating downstream cascades via the JAK/STAT pathway and thereby influencing key biological processes such as apoptosis, proliferation, immune responses and inflammation (Ref. Reference Schwartz, Kanno and Villarino147). In the context of RA, cytokines such as IL-6 bind to IL-6 receptor (IL-6R) on FLS, activating JAK enzymes that subsequently phosphorylate STAT proteins, particularly STAT3 (Ref. Reference Tanaka, Luo and O’Shea148). Once activated, STAT3 translocates to the nucleus, where it induces the expression of numerous pro-inflammatory genes, cytokines and MMPs (Ref. Reference Dutzmann, Daniel and Bauersachs149). This contributes significantly to synovial inflammation, FLS proliferation, invasiveness and, ultimately, joint damage. Given the crucial role of JAK/STAT signalling, pharmacological inhibition of JAK enzymes represents a promising therapeutic strategy for RA. JAK inhibitors, including upadacitinib (JAK1-specific), tofacitinib (JAK1–3), baricitinib (JAK1, JAK2), filgotinib (JAK1) and peficitinib (JAK3) (see Table 2), effectively block JAK enzyme activity, preventing the phosphorylation and activation of STAT3. This interruption halts STAT3 nuclear translocation and reduces the expression of inflammatory mediators and tissue-degrading enzymes (Ref. Reference Tanaka, Luo and O’Shea148). Moreover, preclinical studies have demonstrated that selective targeting of JAK3 can significantly reduce cartilage and bone erosion in animal models of RA, highlighting the therapeutic benefits of pathway-specific interventions (Ref. Reference Milici, Kudlacz and Audoly150). Thus, targeting the JAK/STAT pathway, particularly through STAT3 suppression, offers considerable promise for improving disease management in RA patients by addressing inflammation at the molecular and cellular levels.
Table 2. Summary of drugs for RA and their effects on FLS
MAPK signalling pathway
The MAPK signalling pathway includes extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 as its main family members. This pathway involves various signalling molecules that regulate cellular processes such as differentiation, apoptosis, proliferation and stress responses (Refs. Reference Kim and Choi151, Reference Cargnello and Roux152, Reference Kyriakis and Avruch153). In RA, pro-inflammatory cytokines such as TNF-α, IL-1 and IL-6 can activate ERK, JNK and p38 MAPK (Refs. Reference Schett, Tohidast-Akrad and Smolen154, Reference Thalhamer, McGrath and Harnett155). A cascade of dual-specificity kinases known as MAPK kinases (MKK) induces the phosphorylation of conserved threonine and tyrosine residues, activating MAPK. One study revealed that MKK7 is the most crucial MKK for activating JNKs in RA-FLS (Ref. Reference Inoue, Hammaker and Boyle156).
NF-κB signalling pathway
In RA, FLS play a critical role in signalling pathways that counteract apoptosis by upregulating the expression of anti-apoptotic proteins. Activation of NF-κB also influences FLS proliferation, leading to hyperplasia of FLS in the synovium affected by RA. RANKL, the primary differentiation factor for osteoclastogenesis from myeloid precursors, is upregulated in its expression within the RA synovium due to activated T cells and FLS (Ref. Reference Noss and Brenner11). NF-κB proteins comprise a family of inducible TFs that regulate numerous genes involved in various immune-inflammatory responses. Once activated, NF-κB promotes FLS proliferation and increases its invasion and infiltration capabilities, while simultaneously reducing apoptosis. The NF-κB signalling pathway significantly contributes to the regulation of many genes involved in inflammation, immune responses, and cell proliferation and survival (Ref. Reference Carlsen, Alexander and Austenaa157). The NF-κB signalling pathway consists of NF-κB, NF-κB inhibitor (IκB) and IκB kinases (IKK) (Ref. Reference Hayden and Ghosh158). It has been shown that an IKK inhibitor can effectively reduce the activation and proliferation of RA-FLS without causing cellular toxicity (Ref. Reference Okazaki, Sawada and Nagatani159). Detailed exploration of the complex dynamics of the NF-κB signalling pathway in RA-FLS has been previously documented (Ref. Reference Nejatbakhsh Samimi, Farhadi and Tahmasebi160).
Notch signalling pathway
The Notch signalling pathway is evolutionarily conserved, operating across multiple cell types and developmental stages. It is particularly noted for its role in governing the differentiation of sublining fibroblasts, having been extensively documented to promote the differentiation of mural cells during development. In both humans and mice, there exist Notch ligands, namely delta-like ligand 1 (DLL1), delta-like ligand 3 (DLL3), delta-like ligand 4 (DLL4), Jagged-1 (JAG1) and Jagged-2 (JAG2), along with four Notch receptors (Notch1–4). This signalling pathway is a highly conserved method of intercellular communication and plays a crucial role in cell fate decisions and tissue homoeostasis (Ref. Reference Artavanis-Tsakonas, Rand and Lake161). Its involvement in RA development is well established, functioning downstream of inflammatory signalling to regulate skeletal development and bone remodelling. Recent scRNA-seq analysis of FLS has highlighted the significance of Notch signalling in the pathogenesis of RA, providing a molecular basis for therapeutically targeting stromal cells in RA through the regulation of Notch3 signalling (Ref. Reference Wei, Korsunsky and Marshall162). The expression of Notch3 and Notch target genes is upregulated in RA-FLS. Additionally, studies on Notch3-deficient mice have shown promising potential for RA treatment, potentially alleviating inflammation and joint damage in RA (Ref. Reference Wei, Korsunsky and Marshall162). Based on these findings (Ref. Reference Wei, Korsunsky and Marshall162), an unbiased method called covarying neighbourhood analysis (CNA) has been developed to capture the Notch activation gradient implicated in RA (Ref. Reference Reshef, Rumker and Kang163). This method highlights the role of endothelium-derived Notch signalling in regulating FLS and the resulting inflammation and pathology observed in RA.
IRAK4 signalling pathway
In addition to the direct involvement in joint inflammation and destruction, FLS express various toll-like receptors (TLRs) that activate the IRAK4 signalling pathway. Activation of IRAK4 significantly increases the production of pro-inflammatory cytokines such as IL-6 and MMPs, further promoting inflammation and joint damage (Ref. Reference Ospelt, Brentano and Rengel164). Among the IRAK family members, IRAK4 plays the most critical role due to its ability to initiate downstream inflammatory signals. IRAK4 activates these signals through an essential self-phosphorylation step, without which subsequent signalling molecules remain inactive. Additionally, IRAK4 acts structurally by assembling a multi-protein complex known as the myddosome. This complex, including IRAK1 and MyD88 proteins, stabilizes and amplifies inflammatory signals, ensuring the sustained activation of inflammatory pathways (Ref. Reference Feng, Chen and Shao165). Recent studies have highlighted IRAK4 as a promising therapeutic target. PF-06650833 (zimlovisertib), a small-molecule inhibitor of IRAK4, has demonstrated promising results by effectively reducing cytokine and MMP release in vitro and significantly alleviating arthritis symptoms in animal models (Ref. Reference Winkler, Sun and De166). Currently, zimlovisertib is being evaluated in phase 2 clinical trials, with interim results at week 12 indicating superior clinical efficacy compared to placebo. Furthermore, emerging approaches involving IRAK4 degradation, such as the selective IRAK4 degrader KT-474 (SAR444656), represent innovative strategies aiming to further suppress inflammation by directly reducing IRAK4 protein levels (Ref. Reference Ackerman, Acloque and Bacchelli167). Collectively, targeting the IRAK4 signalling pathway offers substantial therapeutic promise for RA by directly addressing critical inflammatory mechanisms driven by FLS. Such targeted approaches could provide improved disease management, ultimately benefiting RA patients by intervening precisely at the molecular level.
Epigenetic modulations of FLS as a therapeutic strategy
The epigenetic landscape of RA-FLS suggests that therapeutic potential lies in targeting genes or pathways that are differentially regulated. By examining individual epigenetic markers and conducting comprehensive analyses, we can uncover and prioritize genes and pathways that have been previously overlooked but hold promise for drug development. A particularly intriguing approach would be to reshape the RA epigenome, restoring it to a state of normalcy. Although epigenetic alterations are long-lasting, it is feasible to influence the epigenome by targeting the machinery responsible for these modifications, such as histone-modifying enzymes. In animal models of arthritis, several HDAC small-molecule inhibitors have shown promising efficacy. Additionally, givinostat, an oral inhibitor of class I and class II HDACs, has undergone phase 2 trials in treating systemic-onset juvenile idiopathic arthritis and demonstrated moderate clinical efficacy (Ref. Reference Vojinovic, Damjanov and D’Urzo168). While the precise mechanism of action of HDAC inhibitors is largely unknown, they appear to regulate the acetylation status of histones; for instance, givinostat inhibits cytokine production and has anti-inflammatory effects in RA-FLS (Ref. Reference Angiolilli, Kabala and Grabiec169). Moreover, HDAC inhibitors decrease the mRNA level of pro-inflammatory factors such as IL-6 and PTGS2 (Ref. Reference Grabiec, Korchynskyi and Tak170).
Targeting FLS surface markers and novel therapeutic approaches
In addition to signalling pathways, targeted therapy against specific FLS surface receptors has been explored. The potential presence of disease-associated FLS phenotypes can play a role in categorizing illnesses and enable a more precise strategy for dealing with synoviocytes in RA. For instance, current treatments aim at targeting FLS-specific surface receptors, such as CDH11. Notably, mice lacking CDH11 exhibit resistance to inflammatory arthritis. Although a phase 1 trial evaluating the efficacy of RG6125, a monoclonal antibody targeting CDH11, has been completed, subsequent phase 2 trials and further development for treating RA are halted due to inadequate effectiveness (Ref. Reference Finch, Sostelly and Sue-Ling171). The monoclonal antibody ASP5094, which specifically targets integrin alpha-9, abundantly expressed by RA-FLS and contributing to both cell adhesion and inflammation, unfortunately did not yield favourable results in a phase 2a trial. It failed to demonstrate any discernible difference in disease activity scores compared to a placebo (Refs. Reference Emori, Hirose and Ise172, Reference Takeuchi, Tanaka and Erdman173).
Despite this setback, several other promising therapeutic candidates are currently undergoing human trials. Seliciclib, an orally available inhibitor of cyclin-dependent kinases (CDKs), has shown potential in suppressing the proliferation of FLS by inhibiting CDK2 and inducing the CDK inhibitor p21 (Refs. Reference Alessi, Quarta and Savio136, Reference Perlman, Bradley and Liu137). A recent study has reported the findings of a non-randomized, open-label, dose-exploratory phase 1b clinical trial, which aimed to determine the appropriate therapeutic dose, pharmacokinetics and safety of seliciclib, while also providing a preliminary assessment of its potential in targeting the proliferation of FLS for the treatment of RA (see Table 2). The trial involved a total of 37 RA patients who had an inadequate response to TNF inhibitors. Among them, 15 patients were assigned to five different treatment dose groups of seliciclib. The results revealed that the maximum tolerated dose of seliciclib was 400 mg, once daily, with no unexpected safety concerns, thus suggesting the need for further investigation in future studies (Ref. Reference Pratt, Siebert and Cole138).
In the realm of clinical practice, traditional Chinese medicine (TCM) has been used as a therapeutic and supplementary modality for patients in the early stages of RA. Although TCM lacks specific targets, emerging research has highlighted its impact on FLS in RA. For instance, schisandrin, the bioactive component of Schisandra chinensis, inhibits FLS proliferation and invasion, thereby ameliorating RA pathology (Ref. Reference Lin, Liu and Zhang174). Beyond individual drugs, research has also focussed on the anti-arthritis mechanisms of TCM formulas. Qufeng Tongluo formula, a clinical prescription for RA treatment, has shown the ability to inhibit FLS proliferation, migration and invasion in vitro (Ref. Reference Lu, He and Wang175). Additionally, a two-herb formula RL (consisting of Rosae Multiflorae Fructus and Lonicerae Japonicae Flos) has significantly alleviated arthritis in a CIA rat model and attenuated the expression of RANKL in IL-6/sIL-6R-stimulated RA-FLS, which is linked to the STAT3 signalling pathway (Ref. Reference Chen, Wu and Leung176). These findings provide mechanistic insights into targeting FLS as an approach for TCM treatment against RA.
Conclusion
Within autoimmune arthritis, particularly RA, synovial tissue represents a critical area of interest. This review has explored the physiological roles and pathological transformations of FLS and highlighted their significant impact on RA progression. We have further summarized how these cellular alterations shape therapeutic strategies. The metabolic reprogramming and distinct phenotypic changes of RA-FLS have been extensively discussed, emphasizing potential therapeutic targets derived from both immune pathways and bioinformatics analyses. Overall, targeting FLS in RA presents an attractive therapeutic approach, underscoring the need for continued research into both standalone treatments and synergistic combination therapies.
Signalling pathways such as JAK/STAT, MAPK, NF-κB, Notch and IRAK4, along with epigenetic modifications including DNA methylation, histone modifications, SUMOylation and ncRNA regulation, have been established as critical mediators driving the pathological behaviours of RA-FLS. Therapeutic interventions designed to target these pathways offer considerable promise for effective RA management. However, despite significant progress, limitations remain. Current research often faces challenges such as variability in patient response, nonspecific targeting effects and inadequate clinical translation of promising preclinical results. Additionally, RA-FLS exhibit substantial heterogeneity, complicating efforts to design universally effective therapies. Addressing these challenges will require further mechanistic studies, refined patient stratification and innovative therapeutic designs that effectively bridge basic science discoveries with clinical practice.
Author contribution
CL, AL and DH supervised and revised the manuscript. SY, JF and DX wrote and edited the manuscript. CC, ZW, JH, FQ and XY provided their suggestions. All authors contributed to the article and approved the submitted version.
Funding statement
This work is supported by the National Key R&D Program of China (2024YFC3506200 to DH and 2024YFC3506205 to CL), the National Natural Science Foundation Council of China (82472394 and 82172386 to CL, and 82074234 to DH), the 2020 Guangdong Provincial Science and Technology Innovation Strategy Special Fund (Guangdong-Hong Kong-Macau Joint Lab) (2020B1212030006 to AL), the Guangdong Basic and Applied Basic Research Foundation (2022A1515012164 to CL), the Shenzhen Science and Technology Program (JCYJ20210324104201005 and SGDX20240115112400001 to C.L.), the Hong Kong General Research Fund (12102722 and 12106424 to AL), and the Hong Kong RGC Theme-based Research Scheme (T12–201/20-R to AL).
Competing interests
The authors declare none.




 Open access
Open access