



Introduction
Long-chain fatty acid disorders are a group of autosomal recessive inborn errors of metabolism that lead to incomplete oxidation of fatty acids during periods of fasting or physiologic stress, which leads to an energy deficit affecting the heart, skeletal muscle, brain, and liver. The incidence of long-chain fatty acid disorders is less than <1 per 100,000 in the USA, with some variation among countries worldwide.
Reference Marsden, Bedrosian and Vockley1
Mitochondrial trifunctional protein deficiency is a long-chain fatty acid disorder caused by deficiency of mitochondrial trifunctional protein, a multienzyme complex that catalyses the last three steps of mitochondrial long-chain fatty acid
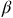 $\beta$
-oxidation.
Reference den Boer, Dionisi-Vici, Chakrapani, van Thuijl, Wanders and Wijburg2
Clinical symptoms of mitochondrial trifunctional protein deficiency range from neonatal onset with severe dilated cardiomyopathy, hypoketotic hypoglycaemia, and liver dysfunction to a later-onset presentation with myopathy, episodic rhabdomyolysis, cardiomyopathy, peripheral neuropathy, and pigmentary retinopathy.
Reference den Boer, Dionisi-Vici, Chakrapani, van Thuijl, Wanders and Wijburg2
Despite newborn screening for these disorders, morbidity and mortality remain high for the neonatal-onset presentations with most infants reported deceased in the first months of life.
Reference Schwantje, Fuchs and de Boer3
$\beta$
-oxidation.
Reference den Boer, Dionisi-Vici, Chakrapani, van Thuijl, Wanders and Wijburg2
Clinical symptoms of mitochondrial trifunctional protein deficiency range from neonatal onset with severe dilated cardiomyopathy, hypoketotic hypoglycaemia, and liver dysfunction to a later-onset presentation with myopathy, episodic rhabdomyolysis, cardiomyopathy, peripheral neuropathy, and pigmentary retinopathy.
Reference den Boer, Dionisi-Vici, Chakrapani, van Thuijl, Wanders and Wijburg2
Despite newborn screening for these disorders, morbidity and mortality remain high for the neonatal-onset presentations with most infants reported deceased in the first months of life.
Reference Schwantje, Fuchs and de Boer3
Treatment of long-chain fatty acid disorders including mitochondrial trifunctional protein deficiency is supportive and includes avoidance of fasting, implementation of diet high in carbohydrates and low in long-chain fatty acids, and supplementation with medium-chain fatty acids. Historically, supplementation has consisted of medium-chain triglycerides, which is comprised of predominantly eight and 10 carbon triglycerides that can bypass the enzymatic defect and provide three molecules of acetyl-CoA for the Krebs cycle. Reference De Biase, Viau and Liu4
Triheptanoin (Dojolvi®) is a synthetic odd-carbon (C7) medium-chain triglyceride composed of three heptanoate (seven carbon fatty acids) molecules linked to a glycerol backbone that has been trialled for use in long-chain fatty acid disorders. Reference Vockley, Enns and Ramirez5 Triheptanoin is metabolised into two molecules of acetyl-CoA and one of propionyl-CoA, thus helping to replenish depleted anaplerotic substrates for the Krebs cycle. Reference Vockley, Enns and Ramirez5 Triheptanoin has been shown to decrease episodes of recurrent rhabdomyolysis, hypoglycaemia, and reverse cardiomyopathy in patients with long-chain fatty acid disorders leading to FDA approval for use in these disorders in 2020. Reference Norris, Scott and Sullivan6
Given the rarity of these disorders, information regarding its use in improving outcomes for neonatal-onset mitochondrial trifunctional protein deficiency has not been published to date. We present a case of neonatal-onset severe mitochondrial trifunctional protein deficiency who presented with acute decompensation with metabolic derangements and severe dilated cardiomyopathy immediately after birth. Triheptanoin was initiated swiftly after diagnosis without significant resolution of dilated cardiomyopathy, suggesting that the efficacy of triheptanoin in such early and severe cases needs to be further investigated.
Case report
The patient was born at 364/7 weeks gestation following a pregnancy complicated by preeclampsia. On the first day of life, the patient developed recurrent hypoglycaemia and increased work of breathing. Chest X-ray revealed cardiomegaly and echocardiography showed severely reduced biventricular function. The patient was intubated, and inotropic support was initiated. The patient deteriorated and was transferred on inotropes to an extracorporeal membrane oxygenation centre where cannulation via the right neck for veno-arterial support was initiated. The patient was transferred on day five of life to our institution for cardiac transplantation evaluation.
Newborn screen collected at 29 h of life resulted on day five of life revealing elevations of long-chain hydroxyacylcarnitines C16-OH 0.94 (RR < 0.16 μmol/L), C16:1-OH 0.28 (RR < 0.26 μmol/L), C18-OH 0.39 (RR < 0.13 μmol/L), and C18:1-OH 0.74 (RR < 0.10 μmol/L), concerning for mitochondrial trifunctional protein deficiency. Plasma acylcarnitines demonstrated markedly elevated concentrations of long-chain 3-hydroxyacylcarnitines despite very low free carnitine and milder elevations of long-chain acylcarnitines, consistent with mitochondrial trifunctional protein. Rapid exome sequencing trio obtained through GeneDx identified a homozygous pathogenic variant c.989del (p.G330Vfs*) in HADHB arising from a uniparental disomy of chromosome 2, confirming the diagnosis of mitochondrial trifunctional protein deficiency.
Treatment was then initiated, including high dextrose intravenous fluids (GIR 13 mg/kg/min), TrophAmine®, and MCT oil at 0.5 mL/hr. Medium-chain triglyceride oil was slowly titrated up to a goal of 30% daily caloric intake. Carnitine was briefly initiated at 20mg/kg but discontinued due to ventricular arrhythmias. After approval, triheptanoin was initiated on day nine of life and titrated to goal dosing of 35% total energy. Reference Norris, Scott and Sullivan6 Continuous enteral feeds via a nasogastric tube, with a small amount of Enfamil™ infant and Vivonex®, were initiated along with triheptanoin.
The patient developed intractable ventricular arrhythmias on day six of life, and balloon atrial septostomy for left atrial decompression was performed. The patient remained on extracorporeal support while receiving triheptanoin and feeds for more than 3 weeks. A wean trial was performed while on inotropic support, which ultimately did not show significant improvement in cardiac function despite adequate nutritional interventions and maximising triheptanoin dosing (Figure 1). Ultimately, the patient was not considered a candidate for ventricular assist device or cardiac transplantation due to the severity of illness and underlying metabolic disease with multi-organ dysfunction. The patient’s multidisciplinary team and parents elected comfort care and discontinued life-sustaining therapies at five weeks of life.

Figure 1. Transthoracic echocardiogram showing severely dilated left ventricle on the apical four chamber ( a ) as well as on the parasternal long axis view ( b ) during extracorporeal membrane oxygenation wean trial.
Discussion
Morbidity and mortality in mitochondrial trifunctional protein deficiency remains high, despite early identification in many patients by newborn screening. Reference Marsden, Bedrosian and Vockley1 Severe disease may present before newborn screen results return, as in our patient. Reference Schwantje, Fuchs and de Boer3 Additionally, newborn screens may miss individuals with long-chain fatty acid disorders. Reference Schwantje, Fuchs and de Boer3 Clinicians should have a low threshold for evaluating newborns and older individuals with cardiomyopathy for these disorders.
In addition to supportive measures and dietary interventions, studies have shown an increased benefit of triheptanoin over the traditional supplementation of medium-chain triglyceride oil in these disorders. Reference Vockley, Enns and Ramirez5 Currently, published studies regarding triheptanoin therapy have shown improvement or reversal of cardiomyopathy in older individuals with long-chain fatty acid disorders, but to our knowledge, there has been no published data regarding initiation of triheptanoin in the neonatal period in patients with severe disease. Reference Vockley, Enns and Ramirez5 While there are rare reports of individuals with mitochondrial trifunctional protein deficiency undergoing cardiac transplantation, they developed cardiomyopathy at older ages and had a less severe disease course than our case. Reference Bursle, Weintraub, Ward, Justo, Cardinal and Coman7
We highlight that despite improvements in therapy and early detection of long-chain fatty acid disorders, mortality and morbidity remain high for these individuals. Our case highlights that the efficacy of triheptanoin in improving the disease course of severe neonatal presentations of long-chain fatty acid disorders needs to be studied further. One potential area of study would be whether earlier implementation of treatment could improve outcomes. In some instances, prenatal diagnosis may be possible either due to known family history or in utero diagnosis of cardiomyopathy, which could present with an opportunity to initiate therapy prior to diagnosis by newborn screening. Reference Spiekerkoetter, Mueller and Cloppenburg8,Reference Bo, Hasegawa and Yamada9 Another area of potential study is ketone body supplementation, as ketone bodies can serve as an alternative source of energy during periods of impaired fatty acid metabolism. Reference van Rijt, Jager and Allersma10 The use of D,L-3-hydroxybutyrate (D,L-3-HB) has been shown to improve cardiac function in patients with multiple acyl-CoA dehydrogenase deficiency and may be a therapy worth investigating further for other fatty acid oxidation disorders. Reference van Rijt, Jager and Allersma10
Acknowledgements
We would like to acknowledge the expert care given by the Pediatric Team at St. Joseph’s Children’s Hospital and the University of Florida ECMO Team, including Mr. Andrew Jaudon, RRT.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Competing interests
None.


 Open access
Open access